Extracellular vesicle miR-93-5p cargo regulates glomerular endothelial cell damage in Alport syndrome
Charmi Dedhia, Valentina Villani, Xiaogang Hou, Paolo Neviani, Geremy Clair, Mohammadreza Kasravi, Cristina Grange, Paolo Cravedi, Paola Aguiari, Velia Alcala, Giuseppe Orlando, Xue-Ying Song, Jonathan E. Zuckerman, Roger E. De Filippo, Stefano Da Sacco, Sargis Sedrakyan

TL;DR
This study shows that miR-93-5p in extracellular vesicles from stem cells can help repair kidney damage in Alport syndrome by restoring endothelial cell function.
Contribution
The study identifies miR-93-5p as a key cargo in extracellular vesicles that mediates their therapeutic effect in Alport syndrome.
Findings
miR-93-5p is downregulated in glomerular endothelial cells during Alport syndrome progression.
hAFSC-EVs restore kidney function by transferring miR-93-5p and regulating VEGFR1 and VEGFR2 signaling.
Spatial transcriptomics confirmed that hAFSC-EVs reverse disease pathways in the glomerulus via miR-93-5p.
Abstract
Modulation of miRNA expression in glomerular cells is associated with renal disease. Here, we investigated the role of miR-93-5p in mitigating glomerular damage in Alport syndrome and whether the disease-modifying activity of extracellular vesicles from human amniotic fluid stem cells (hAFSC-EVs) is mediated by their miR-93-5p cargo. We identified downregulation of miR-93-5p specifically in glomerular endothelial cells in Alport syndrome along disease progression. Silencing of miR-93-5p in hAFSC-EVs changed the transcriptomic and proteomic profile, regulating EV disease-modifying activity. Compared with naive hAFSC-EVs, silenced hAFSC-EVs did not rescue glomerular endothelial function in vitro and did not restore kidney function in vivo. We established that hAFSC-EVs regulate VEGFR1 and VEGFR2 signaling by miR-93-5p cargo transfer, highlighting that miR-93-5p can restore glomerular…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
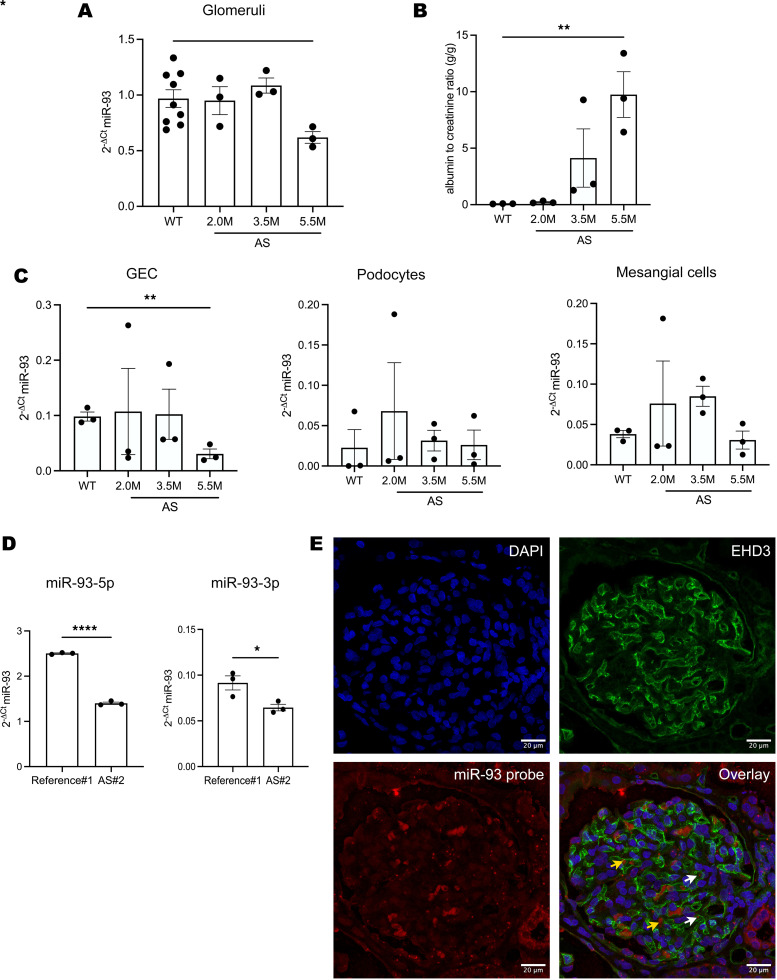 Figure 1
Figure 1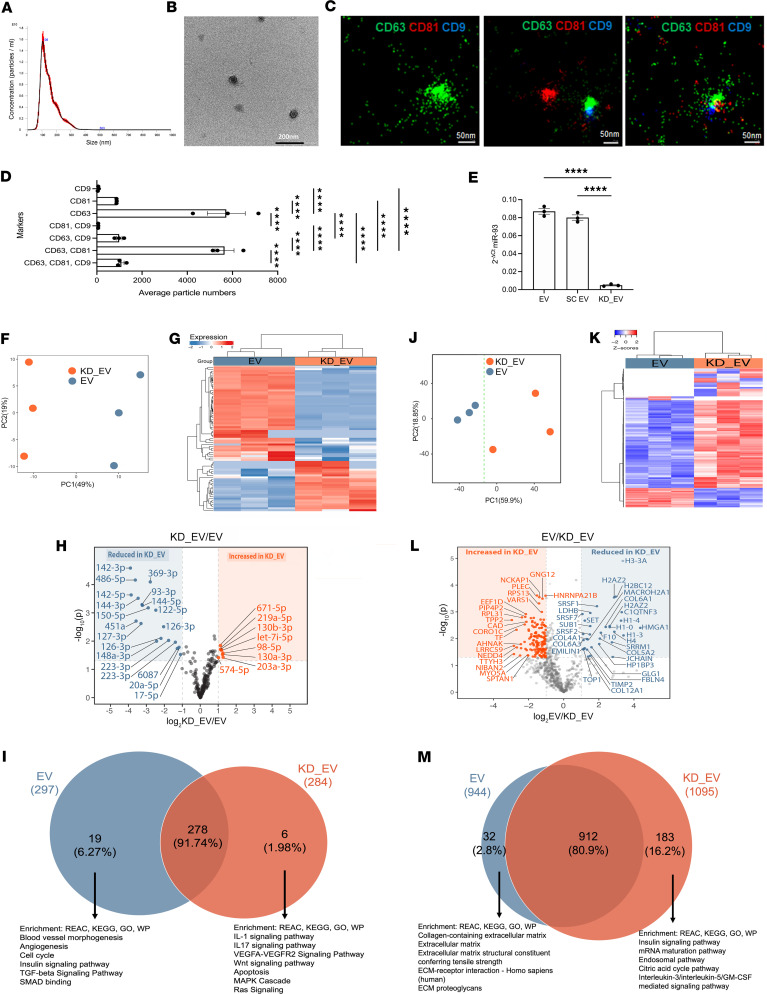 Figure 2
Figure 2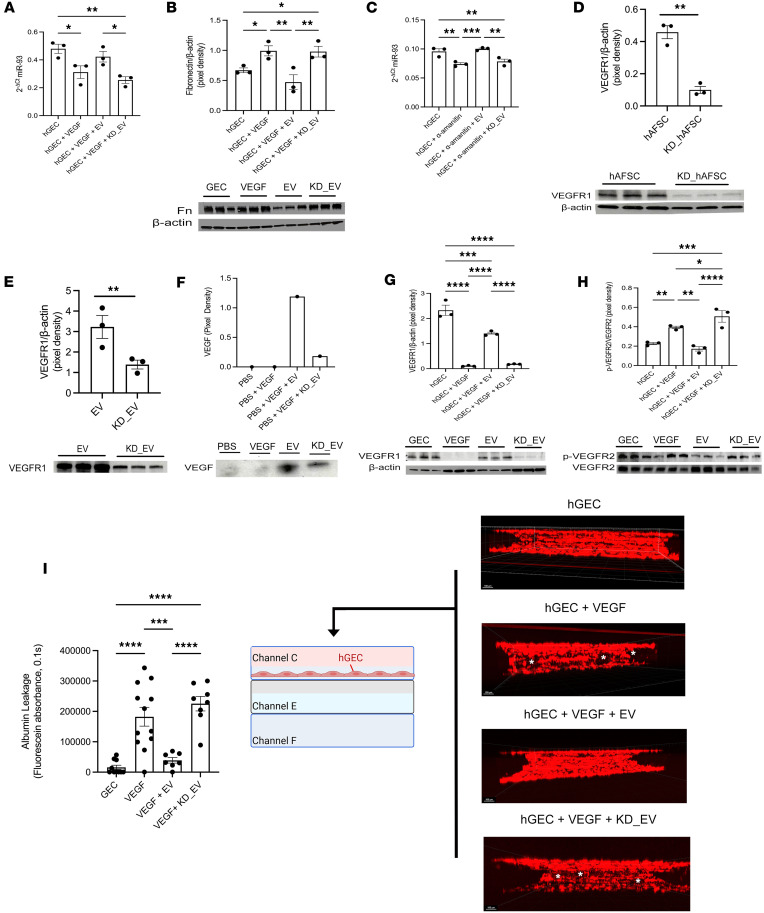 Figure 3
Figure 3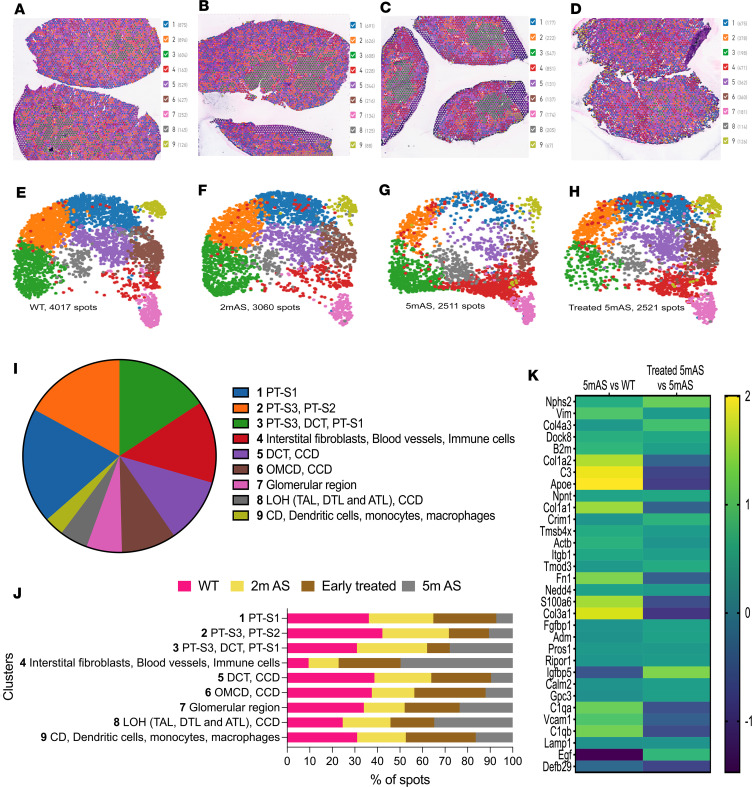 Figure 4
Figure 4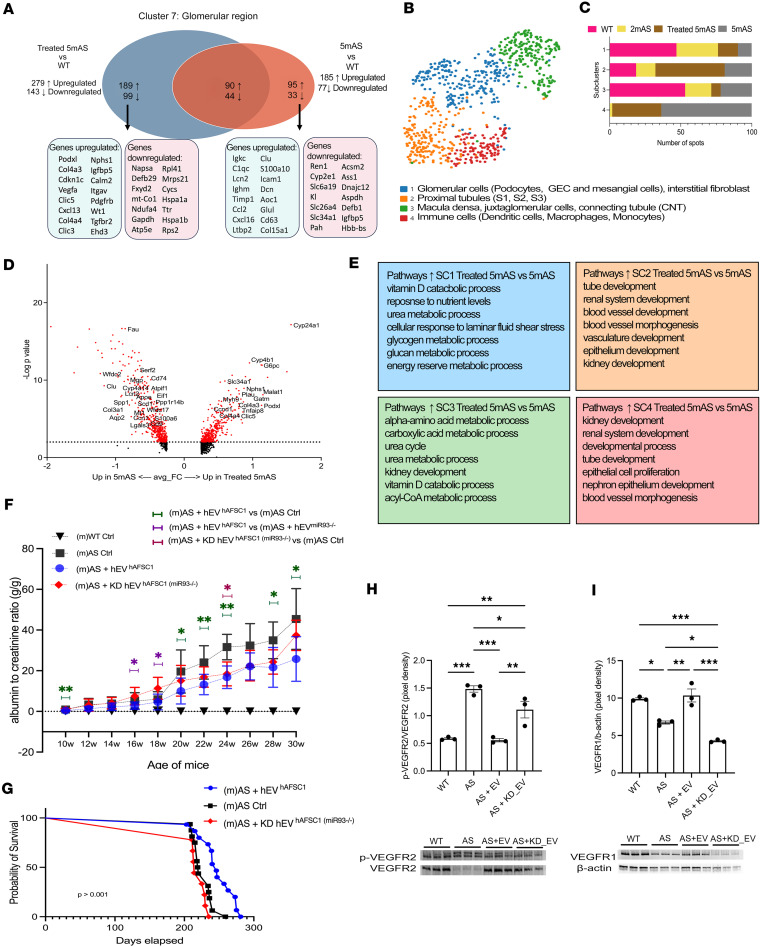 Figure 5
Figure 5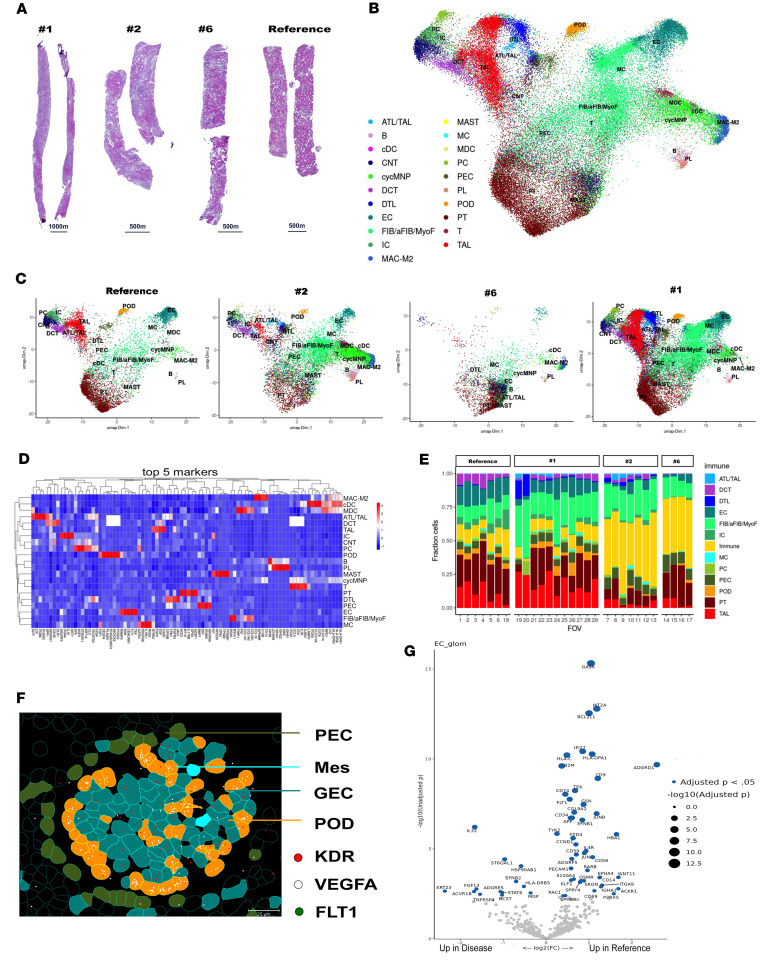 Figure 6
Figure 6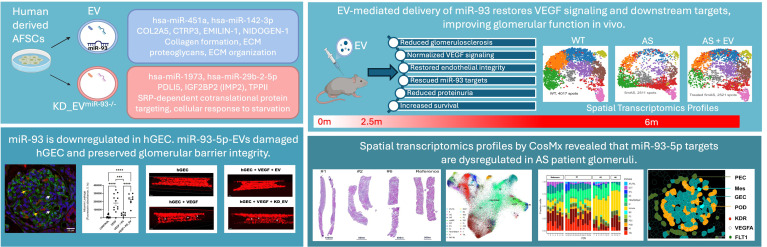 Figure 7
Figure 7- —NIH
- —GOFARR Fund
- —CHLA Extracellular Vesicles Core Pilot
- —CHLA FACS Core Pilot
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCell Adhesion Molecules Research · Extracellular vesicles in disease · Connective Tissue Growth Factor Research
Introduction
Disturbance of the crosstalk between glomerular cells and changes in their interaction with the glomerular basement membrane (GBM) activate disease processes that lead to kidney failure (1). Alport syndrome (AS) is a progressive glomerular disease caused by mutations in the COL4A3, COL4A4, and COL4A5 genes (2, 3), resulting in the improper assembly of the GBM and leading to changes in cell-matrix interactions and altered glomerular cell crosstalk (4–6). Patients with AS, in addition to kidney disease, also present with auditory and ocular symptoms (2, 7, 8). AS management is centered on angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers, and recently on drugs like sparsentan (dual endothelin and angiotensin II receptor antagonist) or sodium-glucose cotransporter 2 inhibitors, which show stronger effects in preserving kidney function (9, 10). Even though these therapies delay the progression to end-stage renal disease, there is an unmet need for new disease-modifying therapies that can directly repair or stabilize glomerular cell biology.
AS is considered a podocyte-centric disease, but we (11–13) and others (14) showed a central role of glomerular endothelial cells (GECs) in AS as well as in other renal diseases. The crosstalk between podocytes and GECs is essential for the function of the filtration barrier (1, 15). For example, GECs are particularly vulnerable to changes in VEGF signaling produced by podocytes (1, 15, 16), as VEGF is a crucial regulator of glomerular capillary homeostasis (17) and it is implicated not only in AS (18) but also in various renal pathologies (1, 16).
Extracellular vesicles (EVs) are fundamental modulators of cell-cell communication (19, 20) and, in particular, stem cell–derived EVs play an important role in modulating molecular pathways ranging from fibrosis to immunomodulation to tissue repair, thus slowing down disease progression in many conditions, including renal disease (21–23). Although their organ-protective role is recognized, the mechanisms involved in the EV-mediated glomerular protection are poorly understood, and further investigation into how EVs function as regulators of cell-signaling dynamics is essential to elucidate their contribution to renal protection.
We previously demonstrated (11) that EVs derived from mouse amniotic fluid stem cells (AFSC-EVs) can modulate VEGF signaling in the glomerulus in AS (Col4a5^–/–^) mice, characterized by a mutation in the α5 chain of collagen IV, the most prominent form of AS in humans (2, 3).
While previous studies using mouse-derived EVs have demonstrated renoprotective effects in experimental models, key questions remain regarding the therapeutic potential of clinically relevant human EV platforms. Therefore, to facilitate the EV clinical translation for chronic kidney disease (CKD), we here studied and characterized human-derived EVs from amniotic fluid.
Among the various components of the EV cargo (proteins, nucleic acids, lipids), microRNAs (miRs), small noncoding RNA molecules (24), are considered key players in exerting EV function; once delivered to the target cells, they modulate gene expression, influencing cell proliferation, differentiation, apoptosis, and other biological processes (25–27).
We previously reported the first evidence that miR-93-5p (hereafter referred to as miR-93), a potent regulator of VEGF signaling (28), plays a key role in GEC biology during AS progression and that miR-93–specific EV cargo transfer is essential for restoring GEC function in AS. Our findings provide insights into the mechanistic role of EVs in regulating GEC biology and emphasize the potential translation of human AFSC-EVs to the clinic as a treatment for patients with AS and other forms of glomerulopathy.
Results
miR-93 expression changes in glomerular cells during AS disease progression.
To study the role of miR-93 in AS, we first determined the expression of miR-93 within the glomeruli of AS versus WT mice along disease progression, showing that miR-93 is downregulated in glomeruli in AS mice at 5.5 months of age (5.5mAS mice) (Figure 1A), when the level of proteinuria is high (Figure 1B). We detected a significant downregulation of miR-93 expression specifically in GECs in AS, but not in podocytes or mesangial cells (Figure 1C and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.197643DS1). miR-93 exists in 2 mature isoforms (miR-93-5p and miR-93-3p); the 3p form is not highly expressed in WT cells and does not change along disease progression in AS cells (Supplemental Figure 2, A–C); therefore, our study focused on the 5p form.
We confirmed that miR-93 expression is downregulated in biopsies of patients with AS and confirmed the biological relevance of the 5p form versus the 3p isoform also in human samples (Figure 1D). Since the mouse data showed that miR-93 is expressed in GECs, we confirmed by in situ hybridization that miR-93 is mainly expressed in GECs also in human glomeruli (Figure 1E and Supplemental Figure 2D). To characterize the glomerular gene signature in AS and to determine whether miR-93 targets are altered in AS glomeruli, we performed bulk RNA-seq analysis on glomeruli isolated from AS mice and age-matched WT mice (Supplemental Figure 3, Supplemental Dataset 1, and NCBI Gene Expression Omnibus [GEO] dataset GSE318476). Principal component analysis (PCA) revealed a clear separation between AS and WT glomeruli (Supplemental Figure 3A), and transcriptional differences were also confirmed by hierarchical clustering (Supplemental Figure 3B). A volcano plot (Supplemental Figure 3C) revealed a significant shift in gene expression in AS versus WT. Specifically, Tnfrsf21 and Tgfbr2, predicted miR-93 targets, were altered in AS glomeruli (Supplemental Figure 3D), thus correlating transcriptional changes in AS with changes in miR-93 targets. Our studies were performed in male AS mice because of the X-linked nature of the disease, and even though heterozygous females were not the focus of our analysis, alteration of miR-93 targets was also present in glomeruli of female AS mice compared with age-matched WT female mice (Supplemental Figure 4, A–D, and Supplemental Dataset 1). Importantly, even if heterozygous females were to present with milder disease with no evident changes in proteinuria during our analysis timeline (Supplemental Figure 4E), they were analyzed to document that their glomerular transcriptome is different versus WT and that miR-93 targets are also modified in the milder form of AS disease.
Loss of miR-93 alters the EV cargo.
miR-93 is one of the most highly expressed miRs in human EVs (Supplemental Dataset 2), with comparable expression in mouse and human EVs (Supplemental Figure 5A). For data reproducibility, we have characterized (and used in all experiments) EVs derived from our established human AFSC (hAFSC) clonal line, as published previously (11, 29–31). The data indicate that in 24 hours, 1 × 10^6^ hAFSCs produced 2.8 × 10^10^ EVs, with a mode size of approximately 108 nm (Figure 2A) and had an intact membrane, as confirmed by TEM (Figure 2B). Super-resolution microscopy confirmed the presence of the tetraspanins CD9, CD63, and CD81, known to be commonly expressed in EVs (19). The majority of EVs were CD63^+^ or CD63^+^CD81^+^, with smaller fractions of CD81^+^, CD63^+^CD9^+^, and CD63^+^CD81^+^CD9^+^ (Figure 2, C and D). We also determine that hAFSC-EVs are CD63^+^VEGFR2^+^, CD63^+^CD81^+^VEGFR2^+^, CD81^+^VEGFR1^+^, CD63^+^VEGFR1^+^, and CD81^+^CD62^+^VEGFR1^+^ (Supplemental Figure 5, B–E).
To understand how miR-93 expression affects EV cargo, we generated miR-93–knockdown EVs (hAFSC-EVs*^miR-93–/–^*, hereafter KD_EVs) using a transient antagomiR method and confirmed silencing in the cells (Supplemental Figure 5F) and EVs (Figure 2E). We performed miR sequencing on naive EVs and KD_EVs (Supplemental Dataset 2). Analysis showed a clear separation between KD_EVs and EVs (Figure 2F), and an overall shift in miR expression between the 2 groups (Figure 2G). A volcano plot indicates that miR-93 KD resulted in significant changes in specific miRs (Figure 2H); the most significantly upregulated and downregulated miRs in KD_EVs are shown. KD of miR-93 resulted in upregulation of miR-1973 and miR-29b-2-5p (only in KD_EVs) and downregulation of miR-4325p, miR-151a-3p, and miR-10b-5p expression in the KD_EVs (Supplemental Dataset 2). Prediction targets of the above-cited miRs include genes that play a key role in CKD progression, like PDGRB, WNK1, TGFb1, VEGF, FBN1, FN, and CADM2 (32–39), with miR-432-5p having the highest predicted target gene, COL4A5. We identified (Figure 2I) the exclusively upregulated miRs in EVs (19 miRs) and KD_EVs (6 miRs). Enrichment analysis of these miRs’ predicted genes in EVs is associated with pathways related to blood vessel morphogenesis, angiogenesis, cell cycle, and the insulin signaling pathway, whereas the analysis of the miR predicted genes in KD_EVs identified pathways such as IL-1 signaling, IL-17 signaling, VEGF and WNT signaling, and apoptosis.
We performed proteomics on the same EVs (Supplemental Dataset 3). Analysis showed that KD_EVs differ from the naive EVs (Figure 2J); protein cargo changes are shown in the hierarchical clustering (Figure 2K) and in the volcano plot (Figure 2L, the most upregulated and downregulated proteins in KD_EVs are shown). From the total identified proteins, 183 were uniquely present in KD_EVs and 32 in the naive EVs (Figure 2M). These uniquely expressed proteins in the KD_EVs are involved in pathways like the insulin signaling pathway, mRNA maturation pathway, and endosomal pathway. Proteins exclusively upregulated in EVs instead displayed enrichment for pathways related to collagen-containing ECM, ECM structural constituents, and cell-matrix adhesion pathways, all important pathways altered during AS progression (Figure 2M). The results from the miR sequencing and the proteomics show that KD of miR-93 expression induces changes in miRs and protein content that are relevant and known to contribute to AS disease.
EVs regulate the miR-93/VEGFR1/VEGFR2 axis in GECs.
Since GECs are the primary source of miR-93 changes along disease progression, we evaluated the effects of EVs or KD_EVs in human GECs (hGECs). Following VEGF-induced damage, miR-93 expression was significantly reduced in hGECs and restored after exposure to EVs but not to KD_EVs (Figure 3A). Fibronectin expression, which has been shown to increase after VEGF-induced damage in hGECs (40) and is also a miR-93 target (28), was restored to normal levels when exposed to naive EVs (Figure 3B). Next, we evaluated whether miR-93 was transferred as part of the EV cargo to hGECs by reducing endogenous miR expression in hGECs using α-amanitin (an inhibitor of miR synthesis, ref. 41). We detected normal expression of miR-93 after treatment with EVs but not with KD_EVs, suggesting a possible direct transfer of miR-93 from EVs to hGECs (Figure 3C).
We previously demonstrated (11) that EVs can reduce the availability of VEGF because of the presence of VEGFR1 (42) on their surface that can trap VEGF. Therefore, we determined whether miR-93 regulates VEGF trapping by modulating the expression of VEGFR1. Silencing of miR-93 reduced expression of VEGFR1 in hAFSCs and EVs (Figure 3, D and E). We performed a coimmunoprecipitation assay using an anti-VEGFR1 antibody and then probed for VEGF, and showed a significantly higher efficiency of EVs in trapping VEGF versus KD_EVs (Figure 3F). We also demonstrated that miR-93 directly regulates VEGFRs in damaged hGECs since KD_EVs do not restore to normal the level of VEGFR1 expression and p-VEGFR2/VEGFR2 activity versus normal EVs (Figure 3, G and H). In addition, albumin leakage assay in the glomerulus/endothelium on-a-chip (GEOAC), generated using the same system as reported previously (43, 44), showed that EVs but not KD_EVs preserve the GEC barrier integrity in VEGF-induced damage (Figure 3I). Taken together, these data confirm that miR-93 modulates the expression of VEGFR1 and the activity of VEGFR2 in GECs and regulates GEC function.
We also investigated the effect of naive EVs versus KD_EVs in human podocytes (Supplemental Figure 6, A and B) and in mesangial cells in vitro (Supplemental Figure 6, C and D). In contrast with GECs and podocytes, damage to mesangial cells induced an upregulation of miR-93, which was restored with KD_EVs and not with normal EVs. This result is surprising but intriguing, as EVs appear to modulate damage in these cells through different mechanisms, possibly not involving a direct miR-93 action. Further investigations focused on understanding EV uptake by different glomerular cells and their cargo’s mechanism of action are needed, and are beyond the scope of this work.
EVs regulate transcriptional changes in vivo.
To correlate changes in histological architecture with changes in gene expression during disease progression and to determine the disease modifying activity of EVs, we used Visium Spacial Transcriptomics (ST) (Supplemental Figure 7, Supplemental Methods, and GEO GSE245039) in WT (4m), 2mAS mice, 5mAS mice, and AS mice injected with hAFSC-EVs at 2.5 months (before high level of proteinuria) and analyzed at 5 months. Histological analysis showed scarring in 5mAS versus 2mAS and WT mice, which was attenuated in injected mice (Supplemental Figure 8, A–D). Each sample was analyzed individually, and spatial maps with cluster annotations and Gene Ontology (GO) analysis are reported in Supplemental Figure 8, E–P, and Supplemental Datasets 4–7.
To note, because of the size and spacing of the ST spot (55 μm spots spaced 100 μm apart), multiple cell types are included within each spot; therefore, each cluster is composed of multiple renal structures. Nevertheless, the cluster identity was confirmed by histological observation and based on the cell-specific genes found elevated within each cluster.
Unsupervised clustering of integrated samples identified 9 clusters (Figure 4, A–H). Cluster annotation and sample contribution per cluster are shown in Figure 4, I and J, and Supplemental Dataset 8, and the most highly expressed genes in each cluster are reported in Supplemental Figure 9). All samples contributed to all the clusters, but there was a clear shift in cluster distribution in 5mAS, with clusters 1, 2, 5, and 6 being overly underrepresented and cluster 4 (immunological and fibrotic interstitium) specifically being overrepresented, whereas the treated and the 2mAS showed similar cluster distribution to the WT (Figure 4J).
Since AS is a disease of the glomerulus, we looked for the transcriptional changes of downstream targets of miR-93 in cluster 7 (the glomerular cluster). We observed upregulation of Fn1 and downregulation of Nphs2, Itgb1, and Vim, which were reversed by human EV administration (Figure 4K). The complete list of the miR-93 targets between the samples is reported in Supplemental Dataset 9. GO analysis of upregulated genes identified regenerative pathways in EV-treated mice versus nontreated mice (Supplemental Figure 10A). Injection of EVs downregulated apoptosis and stimulated changes in metabolic pathways and renal regeneration (Supplemental Figure 10B). A complete list of the pathway analysis is reported in Supplemental Dataset 10. The distribution of differentially expressed genes (DEGs) between EV-treated and non–EV-treated glomeruli compared with WT identified 189 genes that were exclusively upregulated in treated mice (Figure 5A), including Podxl, Cdkn1c, Nphs1, and Wt1, and upregulation of pathways related to VEGF binding, ECM binding, and glomerular development, while the non–EV-treated glomeruli showed upregulation of pathways like misfolded protein, cell death, L13a-mediated translational silencing of ceruloplasmin expression, SRP-dependent cotranslational protein targeting to membrane, pathways also altered in human AS glomeruli, as we published previously (45). These pathways were downregulated in treated mice, showing the potential therapeutic effect of EVs (Supplemental Figure 11A and Supplemental Dataset 11). The distribution of DEGs between EV-treated, non–EV-treated, 2mAS glomeruli versus WT (Supplemental Figure 11B and Supplemental Dataset 12) showed 181 genes exclusively upregulated in treated mice, with enrichment in pathways related to development. A complete list of the pathway analysis is reported in Supplemental Dataset 13.
To deepen our analysis, we reclustered the glomerular spots and identified 4 subclusters (Figure 5B, Supplemental Figure 12, and Supplemental Dataset 14). The enrichment for tubular genes was not surprising based on the resolution of ST, as specified above. Subclusters 1 and 3 were mainly represented by the WT and 2mAS, subcluster 2 by the treated mice, and subcluster 4 almost exclusively by the nontreated and treated AS mice (Figure 5C).
To understand the effects of the EVs in glomerular subclusters, we analyzed the DEGs in treated versus nontreated 5mAS mice (Figure 5D and Supplemental Dataset 15) and identified enrichment of pathways involved in renal development and in lipid metabolism, which were underrepresented in AS (Figure 5E). A complete list of the pathways analysis is reported in Supplemental Dataset 16.
We then injected AS mice before the onset of heavy proteinuria with either EVs or KD_EVs; normal EVs showed significant improvement in proteinuria versus untreated AS mice or mice treated with KD_EVs. Even if mice injected with KD_EVs showed some improvement in proteinuria, they did not increase survival versus mice treated with normal EVs (Figure 5, F and G). We also showed that in vivo, naive EVs can restore to normal the expression of VEGFR1 and the activity of VEGFR2 versus KD_EVs (Figure 5, H and I).
These data confirm that EVs present with disease-modifying activity and that possibly miR-93 is involved in regulating this renoprotection.
miR-93 targets are also altered in GECs of patients with AS.
To translate our data from mouse to human, we applied the CosMx Spatial Molecular Imaging (SMI) platform in biopsies of patients with AS, which allows for the spatial mapping of gene expression at single-cell resolution (46). We studied 3 different patients with AS with known COL4A5 mutations, and we used a biopsy from a transplanted kidney as a reference (Figure 6A and Supplemental Figure 13A). We manually selected 29 fields of view (FOVs), capturing all the glomeruli available (Supplemental Figure 13B). We performed stringent quality control (QC) by eliminating cells with fewer than 20 transcripts (cells that did not pass QC are represented in black in the segmentation analysis). Using the NanoString pipeline and data available (47), we identified 21 cell types (Figure 6, B–D); the mean of the confidence score is shown in Supplemental Figure 13C. Cell deconvolution analysis displayed an abundance of the major glomerular, tubular, and interstitial cells (Figure 6E); a cell type representation for each FOV is shown in Supplemental Figure 14A. Abundances of immune cell types were detected between diseased and nondiseased FOVs, especially in 2 patients with AS, nos. 2 and 6, who both presented arteriolosclerosis, and no. 6 also low-grade IgA nephropathy (Figure 6E; for clarity, the immune cells here were grouped together). We focused on the glomerulus and, using Napari (48), a multidimensional image viewer for Python, we identified the spatial localization of the glomerular cells (Figure 6F, Supplemental Figure 14B, and Supplemental Figure 15). We confirmed that this technology can spatially recognize gene expression at the cellular level by showing that VEGF is expressed in podocytes, and its receptors KDR and FLT1 are expressed in GECs (Figure 6F).
Based on our stringent QC, which reduced the number of cells available for glomerular analysis, we could not quantify gene expression differences for all comparisons for all glomerular cell types/samples. We could confidently only perform analysis on GECs after combining all 3 AS biopsies versus the reference. Despite these limitations, our data confirmed that AS-GECs presented with loss of endothelial markers (PECAM, CD34, and TEK) and showed upregulation of IL32, FGF13, and MPG (Figure 6G). Interestingly, AS-GECs seem to present strong downregulation of the FLT1 gene, which encodes both VEGFR1 and its soluble form sFLT1. Loss of sFLT1 can induce an unbalanced accumulation of VEGF produced by the podocytes, thus causing glomerular damage. These findings confirm target engagement in human AS glomeruli; for example, miR-93 downregulation and FLT1 gene loss were consistently detected in hGECs. Translationally, this suggests that EVs enriched in miR-93 could restore VEGF signaling homeostasis in the human setting. Identifying primary assays critical for characterizing the final EV product in a therapeutic context is paramount for regulatory approval for clinical translation.
Based on our preclinical in vitro and in vivo work and EV characterization derived from an established clonal population of AFSCs, together with the evidence that the major EV targeting signaling (VEGF/miR-93) is present in human AS glomeruli, we can envision a stable set of parameters for our EVs that can guarantee EV lot production with the same characteristics. We can design identity and purity parameters identified by the expression of specific markers, as in Figure 1, but most importantly, we can define a mechanism of action by a potent assay based on the presence of miR-93 and its capability of regulating VEGF signaling. Finally, we can design a disease-modifying activity that can be assessed by lowering proteinuria and by stabilizing the slope of the estimated glomerular filtration rate in patients with AS.
Discussion
EVs are an important cell-to-cell communication system, able to deliver regenerative factors from stem cells to target cells. The mechanisms involved in their therapeutic effects are numerous; they may interact with targeted cells through surface membrane receptors, deliver growth factors present in their corona, or be internalized and act as trophic factors or deliver a specific cargo (19, 49–52). We previously identified that AFSC-EVs could act as decoy factors, trapping VEGF through surface VEGFRs, thus modulating podocyte-GEC crosstalk by attenuating VEGF-induced glomerular injury (11).
In continuing our effort to understand the VEGF-modulating effects of AFSC-EVs in AS, here we characterized the EV cargo, with a specific focus on the miRs, which represent a major component of EV cargo and are modulators of gene expression (19, 53). We previously showed (11) that hAFSC-EVs contain miRs with angio-modulatory properties regulating key components of VEGF signaling, including VEGF (miR-93 and -16.1), VEGF receptors (miR16.1), and different upstream and downstream targets of this signaling (miR-23a, -27a, -221, 322, and -145). Interestingly, miR-93 (miR-93-5p), a key modulator of VEGF signaling (28), compared with all the other angio-modulatory miRs, is highly expressed in our EVs.
Therefore, based on the evidence that miR-93 highly regulates VEGF signaling and that it is highly expressed in EVs, here we investigated whether miR-93 EV–specific cargo transfer would affect EV functional properties in rescuing glomerular injury, since our data showed that miR-93 expression is highly reduced in AS glomeruli and in biopsies of patients with AS.
We first determined that silencing of miR-93 in EVs induced a striking shift in the levels of various miRs (and their targets) and proteins that are critical for glomerular cell function, thus impairing EV renoprotective activity. We observed an upregulation of miR-432-5p, which targets COL4A5 and fibronectin, important for ECM remodeling and AS disease progression (54, 55). To note, even if COL4A5 genes emerged among the predicted miR targets that we identified, in our study, a modulation of this gene might be part of the ECM remodeling process, rather than a direct therapeutic correction. Similarly, miR-10b-5p, which targets ITGb8 (involved in cell-matrix adhesion; ref. 56), was also upregulated. miR-135b-5p, targeting COL4A3, and miR-137, targeting PDGFRa (important for mesangial cell function; ref. 57), were elevated. Concomitantly, we noted a downregulation of key proteins involved in maintaining glomerular architecture and cellular integrity. Loss of miR-93 alters molecular signaling affecting actin polymerization, cadherin binding, collagen trimer formation, and cell-matrix adhesion–related functions, thus suggesting that miR-93 plays a pivotal role in shaping the EV cargo composition and impacts pathways relevant to AS pathogenesis. The importance of miR-93 in regulating AS disease progression was also validated in ST studies. We identified changes in expression of miR-93 targets (like Tgfbr2, Mmp2, Col4a3) specifically in the glomerulus that were restored to normal after EV administration. Most importantly, EVs lacking miR-93 were significantly less efficient in rescuing kidney function in vivo, demonstrating that miR-93 is an important contributor to EV-mediated protection of glomerular homeostasis, although different cargo components (19, 20) and mechanisms likely cooperate in this effect.
The renoprotective role of miR-93 has been reported in other CKDs (58–62) and in an animal model of acute myocardial infarction (63), and it is known that miR-93 directly modulates VEGF expression in podocytes (28) in hyperglycemic conditions.
Interestingly, our data suggest that in AS, miR-93 downregulation is specifically driven by GECs, suggesting a cell-specific mechanism for the dysregulation of miR-93, not involving podocytes or mesangial cells. Mechanistically, we showed that EVs regulate both the expression of VEGFR1 and the activity of VEGFR2 directly in GECs, demonstrating that EVs can modulate VEGF signaling not only by reducing the bioavailability of VEGF produced by podocytes within the glomerular space (11) but also by influencing the receptor activity. Our GEOAC experiments also showed that EVs modulate GEC function by regulating their barrier properties in VEGF-induced damage. Most importantly, we showed that this angio-modulatory effect is regulated by miR-93.
Our studies pointed to miR-93/VEGFR axis regulation in GECs as one possible mechanism of action of EVs and even if it is recognized that EVs induce different effects, our results highlight how disease modulators, like miR-93, might be specific to one cell type in the glomerulus (in GECs at least in AS), since miR-93 expression is not altered in podocytes and mesangial cells during disease progression. These data have important implications for future therapeutics; even if miRs or anti-miRs have been tested in clinical applications (64, 65), including the use of anti–miR-21 (66) in AS, the systemic delivery of naked miRs will not likely be effective in regulating changes specific to one cell type in the glomerulus (or in other cell types). EVs are stable in circulation and protect their cargo, including miRs, from degradation and might reach targeted cells in an effective manner (19, 21, 23, 52). Therefore, we believe that the next generation of EV therapeutics should include disease-specific, cell-targeted EVs, which will allow the generation of effective therapeutic responses where needed.
From a translational perspective, these data highlight how an EV-based therapy might complement current standards of care. By demonstrating that EVs act specifically through miR-93 cargo to restore endothelial VEGF signaling, our findings provide a clear mechanism of action and a measurable marker of target engagement that can guide trial design, while acknowledging that further studies are required to fully establish efficacy, optimize dosing, and clarify the contribution of additional EV cargo.
Our study presents some limitations. ST resolution was limited to multiple cells per spot due to the larger diameter of the spots. Thus, the cell type classification did not achieve single-cell resolution. To overcome this limitation, we subclustered the spots identified as glomerular, and we could evaluate a shift in glomerular cell populations in each sample. We acknowledge the limited number of samples used in the ST studies, but we believe multiple in vivo and in vitro studies support the finding of the ST. Given that KD_EVs failed to reverse damage in our in vitro and in vivo experiments, we did not proceed with a full spatial analysis for the KD_EV condition, but prioritized profiling of untreated AS mice and those treated with hAFSC-EVs at different time points to investigate the baseline disease trajectory and therapeutic potential of EV-delivered miR-93. We recognized the limitations of CosMx; this platform allowed only for 1000 genes (at the time when the analysis was performed), and our QC excluded multiple cells, reducing our ability to analyze DEGs in all cell types. Nevertheless, we used 3 different biopsies with COL4A5 mutations that allowed us to determine changes in human AS GECs. The impact of miR-93 effects within other renal cells remains to be further elucidated, since here we focused on GECs. While the therapeutic effect of the EVs is noticeable in male mice, particularly in our AS colony, it is important to test it also in other AS strains along with female mice.
In summary, we demonstrated that miR-93 exerts key effects on the progression of AS, with GECs emerging as the primary target of miR-93–mediated actions. While our understanding of the mechanism of action of EVs remains to be completed, we suggest that miR-93 plays a predominant role in mediating the EVs’ effects. Based on these collective findings, we believe that EV-based therapies might hold promise as a possible complementary approach to standard-of-care treatment for patients with CKD.
Methods
Sex as a biological variable.
All animal experiments were conducted using male mice since the COL4A5 mutation is an X-linked mutation. In addition, all our previous animal studies were performed in the same male animal model, allowing for direct comparison with existing experiments and thus minimizing variability between datasets. Future animal studies will be necessary to determine whether the results presented here are also evident in heterozygous females and in the Col4a3 and Col4a4 mutations. For human biopsies, we did not consider sex differences and analyzed the biopsies that were available with known COL4A5 mutations.
Animal studies and mouse cell isolation.
AS mice (Col4a5^–/–^; B6.Cg-Col4a5^tm1Yseg^/J) and WT mice (C57BL/6J) were purchased from The Jackson Laboratory and bred as published previously (11). Glomeruli and glomerular cells were isolated as published previously (11), and in vivo experiments are described in Supplemental Methods. The gating strategy for cell isolation is shown in Supplemental Figure 1.
hAFSC culture, EV isolation and characterization, in vitro experiments.
hAFSCs were isolated and cultured as previously described (11, 29–31) and EVs isolated as previously described (11) to avoid bovine EV contamination. EV size, number, TEM, super-resolution microscopy (ONI), EV miR sequencing, and EV proteomics were performed using standard protocols and details are described in the Supplemental Methods. Isolation and culture of human glomerular cells was performed as previously described (43, 44), and protocols for Western blotting, in situ hybridization, and coimmunoprecipitation are reported in Supplemental Methods.
miR-93 quantification, KD_EV generation, in vitro damage, α-amanitin experiments, and GEOAC experiments.
To detect miR-93 expression in mouse samples, the TaqMan miRNA Reverse Transcription kit and the cDNA preamplified with TaqMan PreAmp Master Mix (Thermo Fisher Scientific, 4384267) were used. qPCR was performed using the TaqMan microRNA Assay (Thermo Fisher Scientific). For human samples, total RNA was extracted from paraffin tissue sections using an FFPE RNA Purification Kit (Norgen Biotek Corp., 25400). Small nuclear RNA U6 was used to normalize qPCR results across samples.
KD_EVs were generated by incubating 1 × 10^6^ hAFSCs with miR-93-5p antagomiR (Applied Biological Materials, MNH03941; 1 nmol/10^6^ cells, transient KD) in lipofectamine (Invitrogen, 13778150; 100 μL/10^6^ cells) for 6 hours and EVs isolated as in Sedrakyan et al. (11). The efficiency of miR-93-5p KD in EV cargo was validated by qPCR. To induce cell type–specific damage, 100,000 cells/well were used. GECs were exposed to VEGF (Thermo Fisher Scientific, Phc9394; 400 ng/mL, 3 days), mesangial cells to TGF-β (R&D Systems, 240-B-002; 1 mg/mL, 3 days), and podocytes to puromycin (Sigma-Aldrich, SBR00017; 50 μg/mL, 5 days). To reduce miR expression, hGECs (100,000 cells/well) were incubated with α-amanitin (Sigma-Aldrich, A2263-1MG; 50 μg/mL) with hAFSC-EVs or KD_EVs (1 cell/10^4^ EVs ratio) for 3 hours; 5 μg of total RNA was used to evaluate miRNA expression by RT-PCR.
GEOACs were generated as detailed in the Supplemental Methods. hGECs (30,000 cells) were seeded into the top channel (channel C) inlet at a density of 1.5 × 10^7^ cells/mL. Five days after hGEC seeding, cells were treated with VEGF (400 ng/mL, 6 hours; Thermo Fisher Scientific, Phc9394) to induce injury and with hAFSC-EVs or KD_EVs (1 cell/10^4^ EVs ratio). hGEC barrier function was assessed by albumin absorbance in filtrate collected from channel F as described previously (43, 44).
Transcriptomics studies.
Bulk RNA-seq was performed to determine changes in gene expression and miR-93 targets in AS versus WT glomeruli, while Visium ST (10x Genomics) was used to investigate the renoprotective role of EVs in AS and to determine changes in gene expression along disease progression. SMI (CosMx, NanoString) was performed to evaluate changes in gene expression in a human setting. Detailed description of the procedures and data analysis are described in the Supplemental Methods.
Statistics.
Data are expressed as mean ± SEM. For comparisons between 2 groups, an unpaired 2-tailed Student’s t test was used. For multiple group comparisons, 1-way ANOVA with uncorrected Fisher’s LSD post hoc test was applied unless otherwise specified. Survival curves were analyzed using the log-rank (Mantel-Cox) test. For transcriptomics and proteomics datasets, DEG analysis was performed using DESeq2 (see Supplemental Methods), with an adjusted P value of less than 0.05 considered significant. A P value of less than 0.05 was considered statistically significant throughout. Statistical analysis for the transcriptomics and proteomics data is described in the Supplemental Methods.
Study approval.
AFSCs were obtained as described previously (11, 29–31), and samples were approved by the Children’s Hospital of Los Angeles (CHLA) IRB; exemption was obtained since no consent was required, as samples were deidentified. Samples of amniotic fluid presented with a normal karyotype and confirmed negative for infectious diseases. Discarded kidneys, CHLA IRB approved, were harvested from patients with a non-nephrological cause of death, allowing functional cells for our in vitro studies. For studies of human AS biopsies and healthy tissue biopsies (for Figure 1D, reference no. 1 and AS no. 2; Figure 1E, reference no. 3; and for Figure 6 biopsies are described in B) were obtained from the pathology biorepository at the Department of Pathology and Lab Medicine, David Geffen School of Medicine at UCLA. The IRBs of CHLA and UCLA approved the protocol for the use of archived human samples. All experiments were performed in accordance with ethical guidelines and regulations of the Declaration of Helsinki. Animal studies were performed according to the NIH Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011) and CHLA IACUC approval.
Data availability.
The data supporting the results of this study, including bulk-RNA seq, spatial transcriptomics, and EV analysis cargo, are available from the corresponding author. Raw data and statistics for these experiments are provided in Supplemental Datasets 1–17, Supplemental Methods, Supporting Data Values file, and GEO GSE245039.
Authors contributions
CD performed experiments for all the experiments, analyzed data, and wrote the manuscript. VV analyzed data and wrote the manuscript. XH conducted experiments for glomeruli collection. PN performed EV isolation and characterization. GC performed and analyzed proteomics data. MK conducted GEOAC experiments. CG conducted EV TEM and EV ONI experiments. PC analyzed data and wrote the manuscript. PA conducted cell culture, EV isolation, and data analysis. VA performed in situ hybridization and data analysis. GO provided hGECs and analyzed data. XYS performed ST and analyzed ST data. JEZ provided biopsy and clinical data of patients with AS and references, and revised the manuscript. REDF analyzed data and revised the manuscript. SDS performed GEOAC experiments and performed miR EV sequencing. SS performed bulk RNA-seq and CosMx analyses and revised the manuscript. BB and LP conceived the project, analyzed data, and wrote the manuscript.
Funding support
This work is the result of NIH funding, in whole or in part, and is subject to the NIH Public Access Policy. Through acceptance of this federal funding, the NIH has been given a right to make the work publicly available in PubMed Central.
NIH grant 1R01DK121037-01A1 (to LP).The GOFARR Fund.The Extracellular Vesicles Core Pilot at CHLA.FACS Core Pilot at CHLA.
Supplementary Material
Supplemental data
Supplemental data set 1
Supplemental data set 10
Supplemental data set 11
Supplemental data set 12
Supplemental data set 13
Supplemental data set 14
Supplemental data set 15
Supplemental data set 16
Supplemental data set 17
Supplemental data set 2
Supplemental data set 3
Supplemental data set 4
Supplemental data set 5
Supplemental data set 6
Supplemental data set 7
Supplemental data set 8
Supplemental data set 9
Unedited blot and gel images
Supporting data values
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Fogo AB Mechanisms of progression of chronic kidney disease Pediatr Nephrol 200722122011202210.1007/s 00467-007-0524-017647026 PMC 2064942 · doi ↗ · pubmed ↗
- 2Kashtan CE et al Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport syndrome classification working group Kidney Int 20189351045105110.1016/j.kint.2017.12.01829551517 · doi ↗ · pubmed ↗
- 3Kühn K Basement membrane (type IV) collagen Matrix Biol 199514643944510.1016/0945-053X(95)90001-27795882 · doi ↗ · pubmed ↗
- 4Chew C Lennon R Basement membrane defects in genetic kidney diseases Front Pediatr 201861110.3389/fped.2018.0001129435440 PMC 5796894 · doi ↗ · pubmed ↗
- 5Miner JH The glomerular basement membrane Exp Cell Res 2012318997397810.1016/j.yexcr.2012.02.03122410250 PMC 3334451 · doi ↗ · pubmed ↗
- 6Daehn IS Duffield JS The glomerular filtration barrier: a structural target for novel kidney therapies Nat Rev Drug Discov 2021201077078810.1038/s 41573-021-00242-034262140 PMC 8278373 · doi ↗ · pubmed ↗
- 7Jais JP et al X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males J Am Soc Nephrol 200011464965710.1681/ASN.V 11464910752524 · doi ↗ · pubmed ↗
- 8Savige J et al Genotype-phenotype correlations for pathogenic COL 4A 3-COL 4A 5 variants in X-linked, autosomal recessive, and autosomal dominant Alport syndrome Front Med (Lausanne)2022986503410.3389/fmed.2022.86503435602506 PMC 9120524 · doi ↗ · pubmed ↗