Comprehensive In Silico Investigation of L-Glutamine Transporters and Metabolism in Glioblastoma
Sachin Kumar, Chih-Yang Wang, Helena Kishore Lalwani, Juan Lorell Ngadio, Fitria Sari Wulandari, Daniel Dahlak Solomon, Hui-Pu Liu

TL;DR
This study identifies key metabolic regulators linked to glutamine in glioblastoma, offering potential targets for treatment.
Contribution
The paper introduces three novel glutamine-associated metabolic regulators with clinical relevance in glioblastoma.
Findings
Ceruloplasmin (CP) is associated with redox regulation and poor clinical outcomes in GBM.
SLC25A13 is linked to mitochondrial metabolite exchange and nucleotide biosynthesis.
SLC38A2 is connected to glutamine uptake and growth signaling in GBM.
Abstract
Background/Objectives: Glioblastoma (GBM) is the most aggressive primary brain tumor in adults and remains associated with poor prognosis despite multimodal therapy. Metabolic reprogramming, particularly increased dependence on glutamine, supports GBM bioenergetic, biosynthetic, and redox demands. This study aimed to systematically identify glutamine-associated metabolic regulators with prognostic relevance and biological plausibility in GBM. Methods: Transcriptomic data from TCGA and GTEx were analyzed using GEPIA2, with survival validation performed using the CGGA. Functional pathway enrichment, protein expression assessment, protein–protein interaction network analysis, tumor microenvironment evaluation, epigenetic profiling, and single-cell RNA sequencing validation were integrated to contextualize candidate genes. Pharmacogenomic correlation analysis and structure-based molecular…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1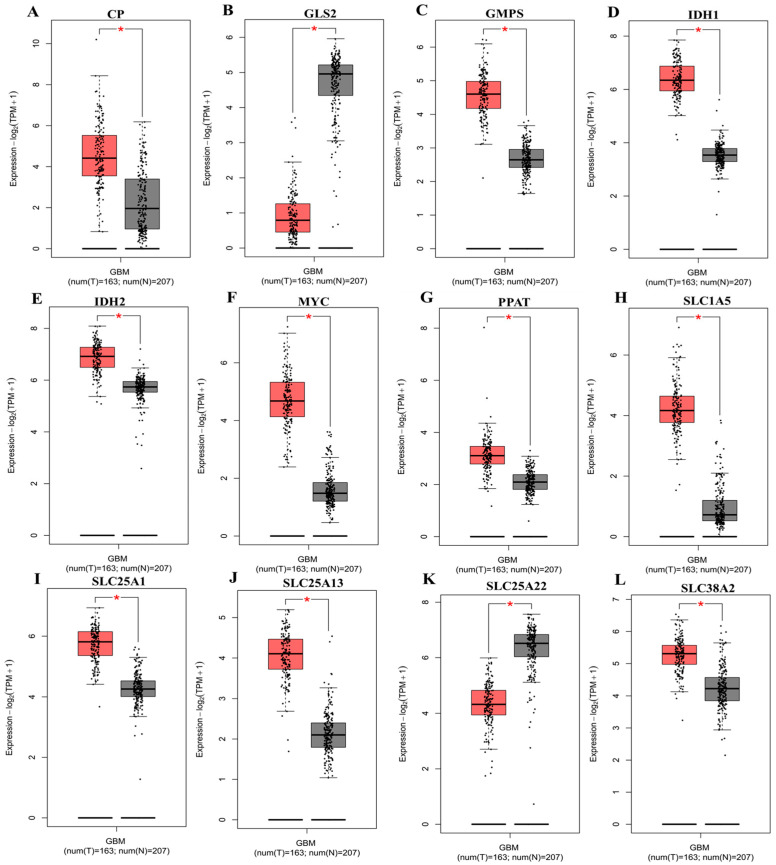 Figure 2
Figure 2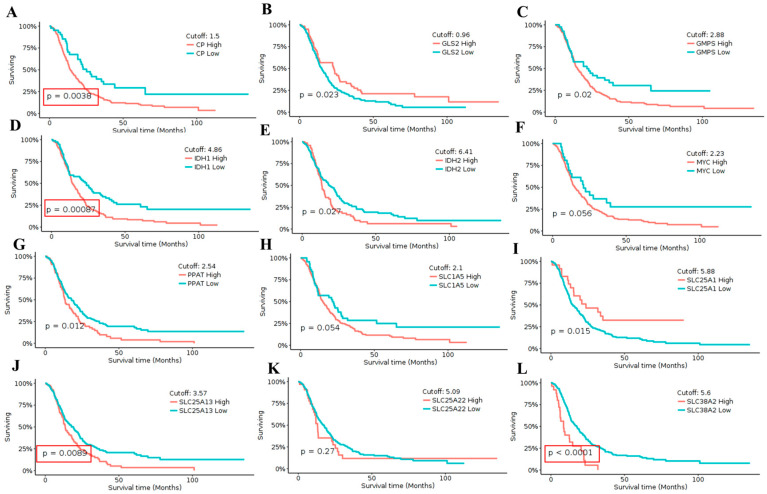 Figure 3
Figure 3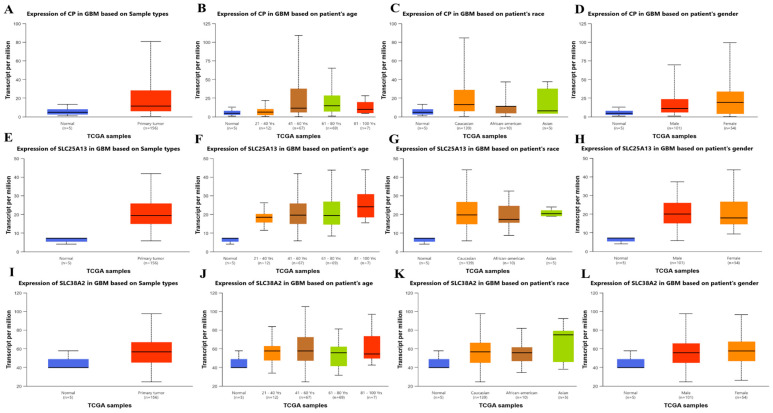 Figure 4
Figure 4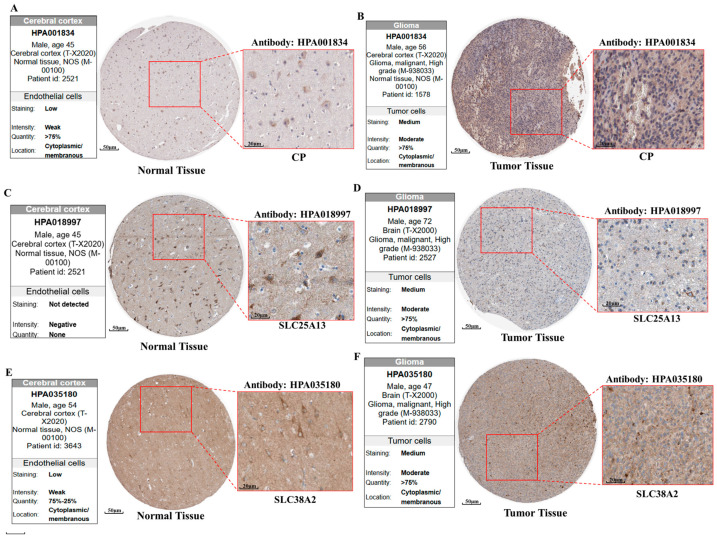 Figure 5
Figure 5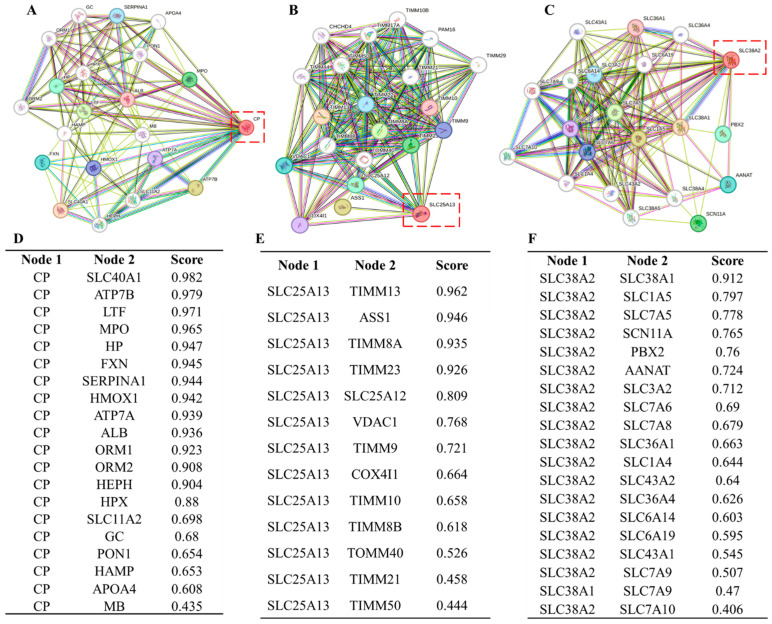 Figure 6
Figure 6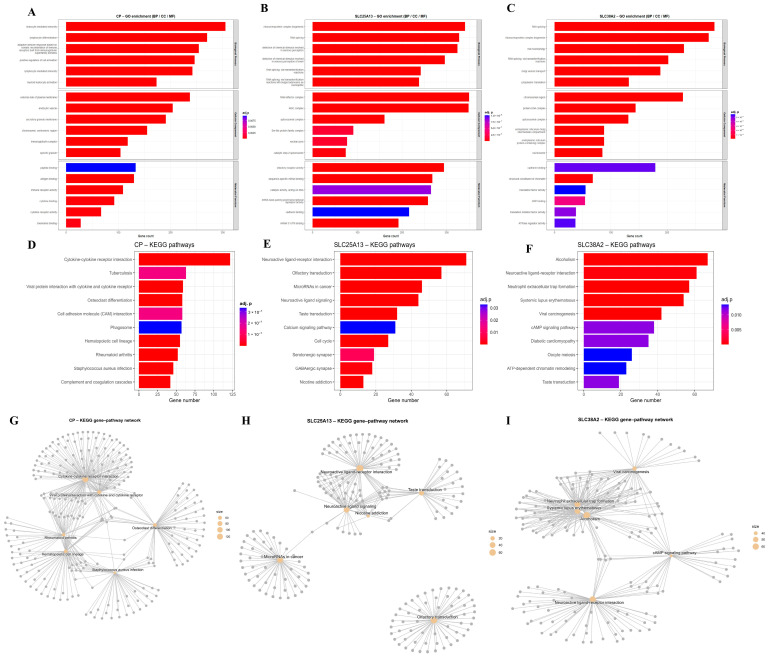 Figure 7
Figure 7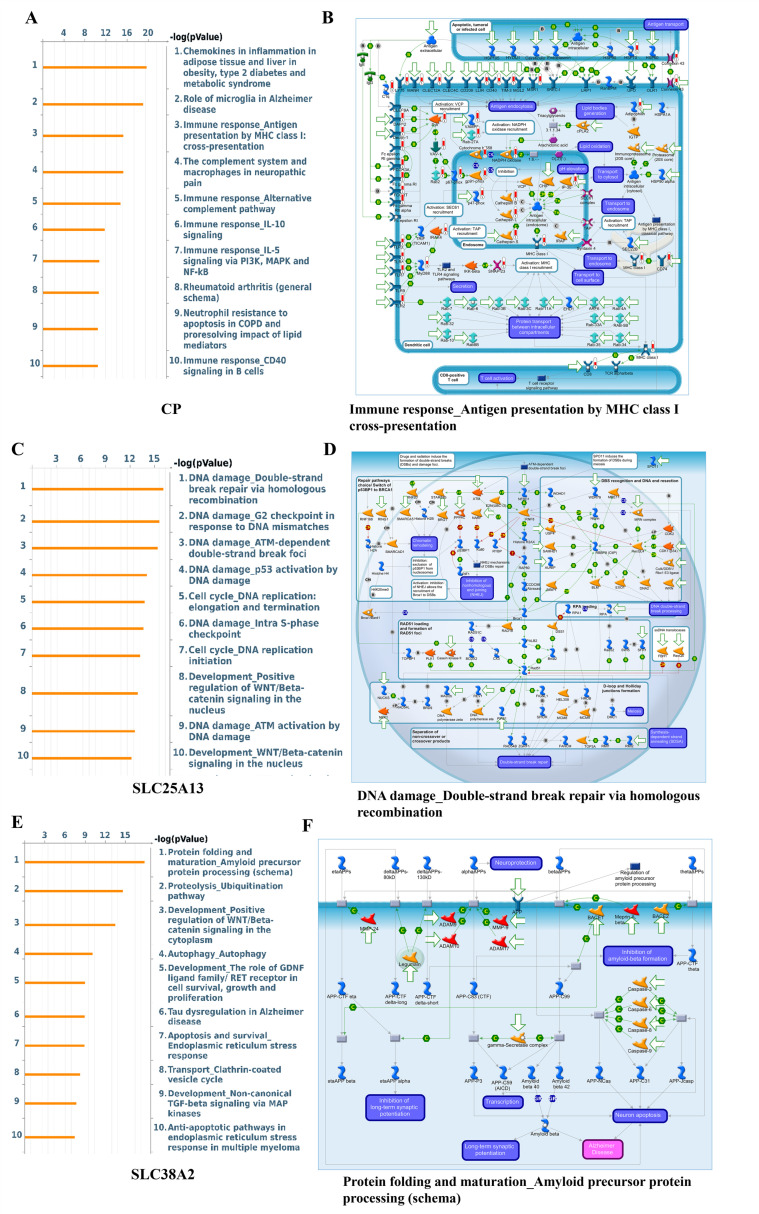 Figure 8
Figure 8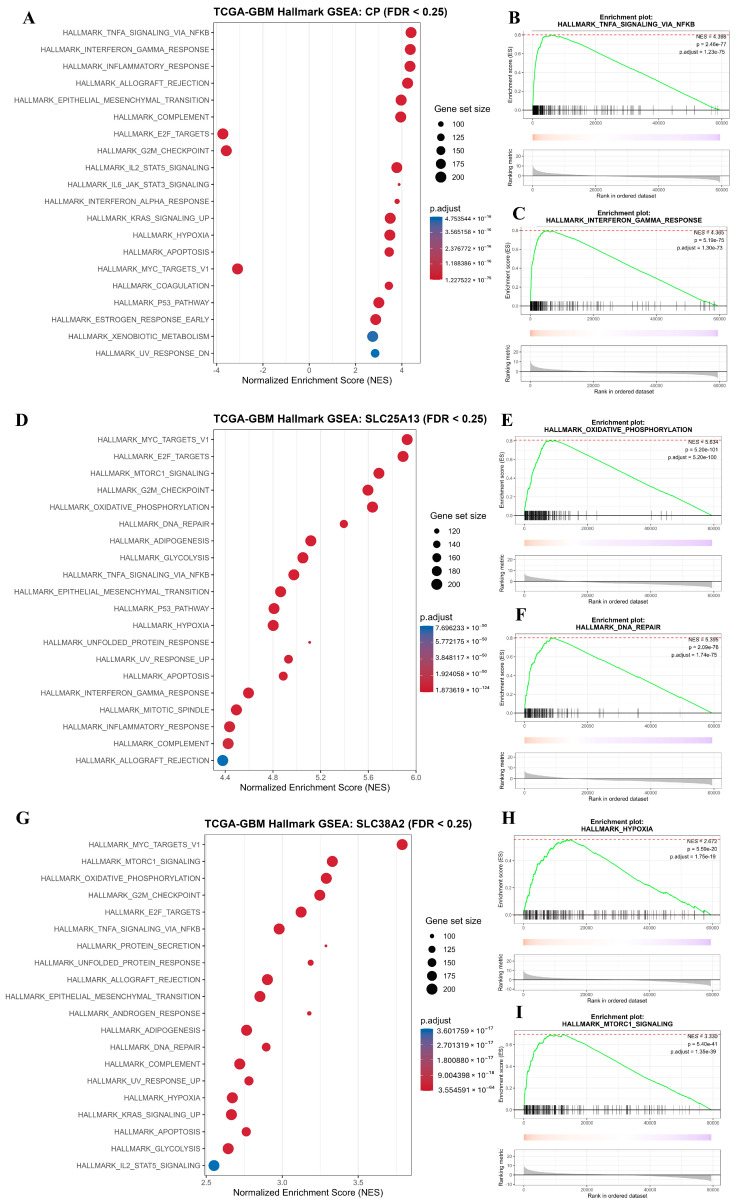 Figure 9
Figure 9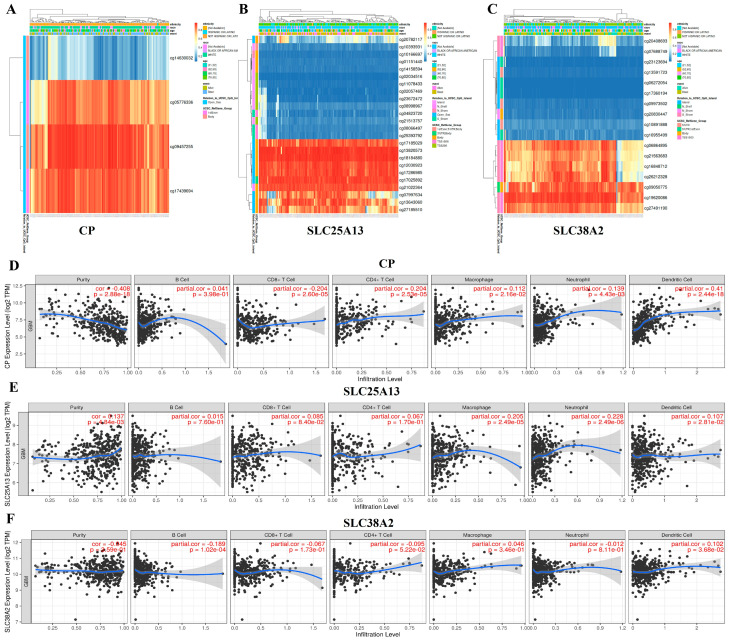 Figure 10
Figure 10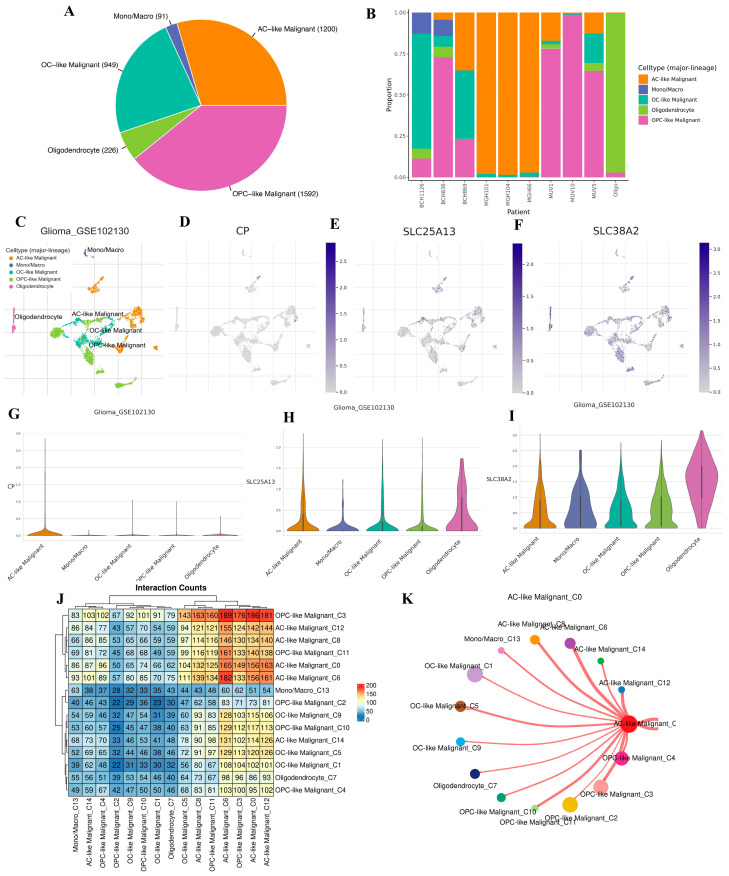 Figure 11
Figure 11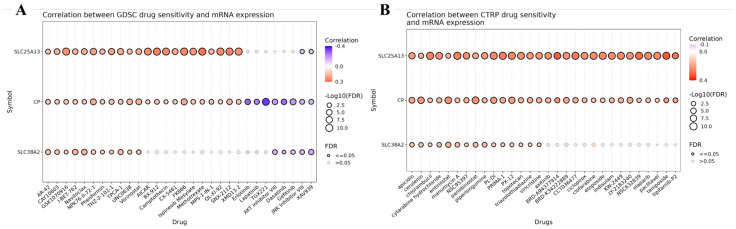 Figure 12
Figure 12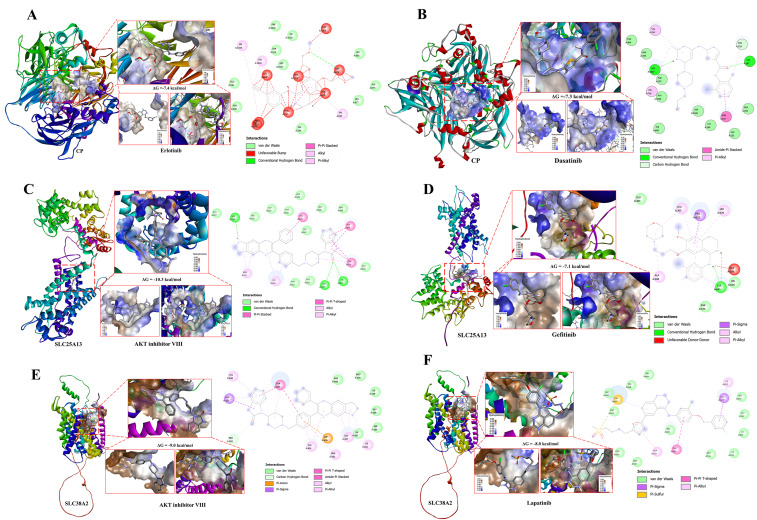 Figure 13
Figure 13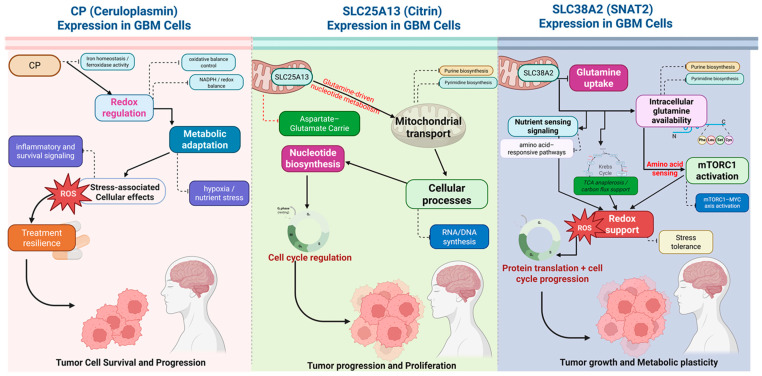 Figure 14
Figure 14- —Kaohsiung Armed Forces General Hospital
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer, Hypoxia, and Metabolism · Amino Acid Enzymes and Metabolism · Ferroptosis and cancer prognosis
1. Introduction
Glioblastoma (GBM) represents the most aggressive form of primary brain cancer in adults and remains largely incurable despite decades of therapeutic advances [1,2]. The limited clinical efficacy of current standard-of-care treatments reflects not only the genetic heterogeneity of GBM but also its extraordinary capacity for metabolic adaptation. Tumor cells within the brain must survive fluctuating nutrient availability, hypoxia, and oxidative stress, requiring continuous rewiring of core metabolic programs. Understanding how GBM sustains these metabolic states is therefore central to identifying biologically meaningful vulnerabilities [3,4].
Amino acid metabolism has emerged as a critical determinant of tumor fitness, with glutamine occupying a central role in cancer cell survival [5]. Glutamine supports cellular proliferation by providing carbon for mitochondrial metabolism, nitrogen for nucleotide and amino acid biosynthesis, and reducing equivalents for redox homeostasis. While glutamine is dispensable for most normal cells, many cancers exhibit a heightened dependence on extracellular glutamine, reflecting a shift toward anabolic metabolism [6]. In GBM, this dependency is particularly pronounced due to the high energetic demands imposed by rapid proliferation and adaptation to the metabolically constrained brain microenvironment. Rather than relying on a single metabolic reaction, glutamine utilization in cancer is governed by an integrated network of transporters, mitochondrial carriers, and metabolic enzymes that collectively regulate glutamine uptake, intracellular trafficking, and metabolic flux distribution. Plasma membrane transporters facilitate glutamine entry into tumor cells, while mitochondrial carriers enable efficient coupling of glutamine-derived metabolites to the tricarboxylic acid cycle, nucleotide synthesis, and redox balance. This coordinated regulation allows tumor cells to dynamically allocate glutamine toward biosynthetic or bioenergetic pathways in response to environmental stress. In GBM, glutamine metabolism is further shaped by oncogenic signaling pathways and transcriptional programs that reinforce metabolic dependency. However, emerging evidence suggests that glutamine-associated metabolic reprogramming is not uniform across all related genes. Instead, GBM appears to selectively amplify specific transport and exchange nodes that support tumor growth while suppressing others that may be incompatible with the malignant state. Although well-characterized metabolic drivers such as IDH mutations have provided important insights into glioma biology, they do not fully explain the broader metabolic landscape of GBM, nor do they capture transport-centric mechanisms that regulate glutamine availability and utilization [6,7,8].
Despite the recognized importance of glutamine metabolism in GBM, a systematic evaluation of glutamine-associated genes integrating transcriptional deregulation, clinical relevance, pathway context, epigenetic regulation, tumor cell specificity, and functional perturbability remains limited. In particular, glutamine transporters and mitochondrial carriers have been relatively underexplored as biomarkers and potential metabolic control points, even though they occupy key positions at the interface between nutrient availability and intracellular metabolic programs [7]. In this study, we conducted an integrative in silico analysis to comprehensively characterize glutamine-associated metabolic regulators in GBM. Using transcriptomic data from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx), we identified genes exhibiting tumor-specific expression patterns, followed by survival analysis using Chinese Glioma Genome Atlas (CGGA) to determine clinical relevance. Functional pathway enrichment, protein-level validation, epigenetic profiling, tumor microenvironment analysis, and Single-cell RNA sequencing (scRNA-seq) were employed to contextualize candidate genes within GBM biology. Finally, pharmacogenomic correlation analysis and structural modeling were incorporated as supportive validation layers to assess the feasibility of perturbing these metabolic nodes. Through this multi-dimensional framework, we aim to identify glutamine-associated regulators that reflect dominant metabolic states in GBM and may serve as biologically meaningful biomarkers and candidates for future metabolic targeting strategies.
2. Results
2.1. Study Design and Transcriptomic Analysis of Glutamine-Associated Genes in Glioblastoma
To systematically investigate glutamine-associated metabolic alterations in GBM, we adopted an integrated in silico study design combining transcriptomic profiling with biological and clinical prioritization. Publicly available RNA-sequencing data from TCGA and GTEx project were utilized to capture tumor-specific transcriptional changes while minimizing confounding effects from normal brain physiology. This strategy enabled the identification of metabolic genes that are selectively deregulated in GBM. Based on an extensive literature-guided framework focusing on glutamine transport, mitochondrial metabolism, and nucleotide biosynthesis, a focused list of candidate genes was defined. This list included CP, GLS2, GMPS, IDH1, IDH2, MYC, PPAT, SLC1A5, SLC25A1, SLC25A13, SLC25A22, and SLC38A2, representing key nodes linking glutamine utilization to biosynthetic and proliferative pathways (Figure 1).
To further evaluate transcriptional deregulation of these genes, differential expression analysis was performed using the GEPIA2 platform. Gene expression levels were compared between GBM tumor samples and normal brain tissues derived from TCGA and GTEx datasets. Several genes demonstrated significant tumor-associated upregulation, including CP, GMPS, MYC, PPAT, SLC1A5, SLC25A1, SLC25A13, and SLC38A2, indicating activation of glutamine transport, mitochondrial metabolite exchange, and nucleotide biosynthesis programs in malignant cells (Figure 2A–L). In contrast, GLS2, and SLC25A22 displayed relatively higher expression in normal brain tissue, highlighting that GBM does not globally upregulate all components of glutamine metabolism but instead exhibits selective pathway rewiring.
To determine the clinical relevance of these transcriptional alterations, survival analysis was subsequently performed using GlioVis. High expression of CP was significantly associated with poorer overall survival (p = 0.0038), indicating a strong adverse prognostic impact (Figure 3A). Similarly, elevated expression of GMPS (p = 0.02), IDH1 (p = 0.00087), PPAT (p = 0.012), SLC25A1 (p = 0.015), SLC25A13 (p = 0.0089), and SLC38A2 (p < 0.0001) was associated with significantly reduced survival, supporting their potential involvement in aggressive disease behavior (Figure 3C,D,G,I,J,L). In contrast, MYC (p = 0.056), SLC1A5 (p = 0.054), and SLC25A22 (p = 0.27) did not reach statistical significance, indicating limited prognostic contribution in this cohort (Figure 3F,H,K). Among the genes showing the strongest and most consistent survival associations, CP, IDH1, SLC25A13, and SLC38A2 emerged as the most statistically significant candidates based on both expression and outcome analyses (Figure 2A–L and Figure 3A–L). Although IDH1 demonstrated a highly significant survival association, it was not pursued further due to its well-established and extensively characterized role as a canonical glioma biomarker, largely driven by mutation-dependent mechanisms rather than transcriptional regulation (Figure 3D).
Based on the convergence of tumor-specific upregulation and robust adverse survival associations, CP, SLC25A13, and SLC38A2 were selected for further study. This selection strategy prioritized relatively underexplored metabolic and transport-associated nodes within glutamine metabolism that may represent clinically relevant and targetable vulnerabilities in GBM.
2.2. Integrated Clinical, Molecular, and Network Characterization of CP, SLC25A13, and SLC38A2 in GBM
Following transcriptomic prioritization, clinical relevance, protein-level expression, and functional network integration of CP, SLC25A13, and SLC38A2 were systematically examined to establish their biological roles in GBM. Expression stratification across clinicopathological variables demonstrated that all three genes exhibit consistently higher transcript levels in primary GBM tumors compared with normal brain tissue, with variable distributions across patient age, race, and gender categories (Figure 4A–L). The predominance of tumor-associated expression across all strata indicates that transcriptional activation of these genes is driven primarily by malignant transformation rather than demographic factors. From a biological perspective, this pattern is consistent with GBM-associated metabolic stress. CP is closely linked to iron metabolism and redox homeostasis, processes that are critical in hypoxic and oxidative tumor microenvironments. SLC25A13, a mitochondrial aspartate-glutamate carrier, plays a central role in coupling mitochondrial metabolism with cytosolic nucleotide biosynthesis by facilitating nitrogen and carbon flux. SLC38A2, a sodium-coupled neutral amino acid transporter, supports adaptive amino acid uptake under nutrient-limited conditions, a hallmark of highly proliferative GBM cells.
Protein-level validation further supported the functional relevance of these transcriptional changes. Immunohistochemical analysis revealed weak or absent staining of CP, SLC25A13, and SLC38A2 in normal cerebral cortex, whereas GBM tissues displayed increased staining intensity and broader cellular positivity (Figure 5A–F). Predominantly cytoplasmic and membranous localization patterns were observed, consistent with the established transport and metabolic functions of these proteins. The concordance between mRNA upregulation and protein expression indicates active engagement of these pathways at the functional level rather than passive transcriptional dysregulation.
To further contextualize their biological roles, protein–protein interaction network analysis was performed. CP demonstrated strong connectivity with proteins involved in iron handling, oxidative stress response, and inflammatory regulation, suggesting integration into redox-sensitive metabolic networks that support tumor survival under oxidative pressure (Figure 6A,D). SLC25A13 showed extensive interactions with mitochondrial transport machinery and metabolic enzymes, highlighting its role as a hub coordinating mitochondrial–cytosolic metabolite exchange and biosynthetic flux (Figure 6B,E). SLC38A2 exhibited broad connectivity with amino acid transporters and metabolic signaling components, consistent with its function in maintaining amino acid availability and metabolic plasticity in GBM cells (Figure 6C,F).
Collectively, the convergence of clinicopathological expression patterns, protein-level activation, and central positioning within metabolic interaction networks underscores the biological relevance of CP, SLC25A13, and SLC38A2 in GBM (Figure 4A–L, Figure 5A–F and Figure 6A–F). These integrated features support their selection as key glutamine-associated metabolic nodes for subsequent functional enrichment, prognostic refinement, and therapeutic exploration.
2.3. Integrated Functional and Pathway-Level Characterization of CP, SLC25A13, and SLC38A2 in GBM
To elucidate the biological programs associated with CP, SLC25A13, and SLC38A2, functional enrichment analyses were performed to map their correlated gene sets to Gene Ontology (GO), KEGG pathways, curated signaling maps, and Hallmark gene sets. Collectively, these analyses reveal a convergent activation of metabolic, biosynthetic, stress-response, and proliferative programs that are central to GBM progression.
GO enrichment demonstrated that genes positively correlated with CP, SLC25A13, and SLC38A2 are predominantly involved in biological processes related to metabolic regulation, cellular stress adaptation, biosynthetic activity, and cell cycle control, indicating that these genes are embedded within high-demand cellular states characteristic of malignant growth (Figure 7A–C). At the pathway level, KEGG analysis revealed enrichment of metabolic and signaling pathways linked to nucleotide metabolism, amino acid transport, oxidative phosphorylation, DNA damage response, and cancer-associated signaling cascades, underscoring the integration of glutamine-related transport and metabolism with proliferative control mechanisms (Figure 7D–F). Network-based visualization of enriched gene sets further highlighted the extensive connectivity of CP-, SLC25A13-, and SLC38A2-associated genes, forming densely interconnected modules rather than isolated functional clusters (Figure 7G–I). This network topology supports the concept that these genes act as central coordinators within broader metabolic and regulatory systems, rather than functioning through single linear pathways. SLC25A13-centered networks emphasize mitochondrial transport and metabolic coupling, while SLC38A2-centered networks are enriched for amino acid transporters and nutrient-sensing components, reflecting adaptive responses to metabolic stress.
To refine pathway-level interpretation, curated signaling maps were examined using MetaCore analysis. CP-associated gene sets showed enrichment of inflammatory and immune-modulatory signaling, including chemokine- and cytokine-driven pathways, consistent with CP’s established role in redox balance and iron-associated inflammatory responses within the tumor microenvironment (Figure 8A). In contrast, SLC25A13-correlated genes were strongly associated with DNA damage response, cell cycle checkpoint regulation, and replication stress pathways, including ATM/ATR signaling and S-phase control, indicating a tight coupling between mitochondrial metabolite transport and genome maintenance during rapid cell division (Figure 8B). SLC38A2-associated networks were enriched for protein folding, stress-response signaling, and growth factor–driven pathways, reflecting its role in sustaining amino acid availability under conditions of nutrient limitation and oncogenic stress (Figure 8C).
Consistent with these findings, Hallmark gene set enrichment analysis revealed coherent transcriptional programs associated with high expression of each gene. CP expression correlated with enrichment of inflammatory signaling pathways, including TNFα signaling via NF-κB, interferon responses, epithelial–mesenchymal transition, and cell cycle–associated gene sets, indicating a link between metabolic stress, inflammation, and invasive tumor phenotypes (Figure 9A–C). Elevated SLC25A13 expression was associated with strong enrichment of MYC targets, E2F targets, oxidative phosphorylation, DNA repair, and G2M checkpoint pathways, highlighting a transcriptional state driven by high biosynthetic demand and replication stress (Figure 9D–F).
Similarly, SLC38A2 expression correlated with enrichment of MYC signaling, mTORC1 signaling, hypoxia, and cell cycle progression pathways, reflecting adaptive metabolic rewiring that supports proliferation under hypoxic and nutrient-deprived conditions (Figure 9G–I).
Across all analyses, a coherent biological theme emerges: CP, SLC25A13, and SLC38A2 converge on transcriptional and signaling programs that integrate glutamine-associated transport and metabolism with inflammation, DNA damage tolerance, cell cycle progression, and stress adaptation. These enrichments represent correlated expression states characteristic of aggressive GBM biology rather than direct evidence of pathway activation or regulatory control by individual genes. Nevertheless, the consistency across GO, KEGG, interaction networks, curated signaling maps, and Hallmark gene sets supports the concept that these three genes occupy central positions within glutamine-driven metabolic and proliferative networks, providing a functional foundation for their prioritization in downstream mechanistic and therapeutic analyses (Figure 7A–I, Figure 8A–C and Figure 9A–I).
2.4. Epigenetic Regulation and Immune Microenvironment Associations of CP, SLC25A13, and SLC38A2 in GBM
To investigate epigenetic mechanisms underlying the dysregulated expression of CP, SLC25A13, and SLC38A2 in GBM, DNA methylation profiles were examined across tumor samples. Gene-centric methylation heatmaps revealed heterogeneous but gene-specific CpG methylation patterns, reflecting distinct epigenetic architectures rather than uniform methylation shifts across the genome (Figure 10A–C). For each gene, CpG probes clustered into hypomethylated and hypermethylated regions, indicating selective epigenetic remodeling in GBM. For CP, differential methylation across CpG sites suggests partial promoter and gene-body hypomethylation in a subset of tumors, a configuration frequently associated with transcriptional activation of stress-responsive and metabolic genes (Figure 10A). Such epigenetic relaxation may facilitate sustained CP expression under oxidative and inflammatory conditions characteristic of the GBM microenvironment. SLC25A13 exhibited pronounced hypomethylation at CpG loci linked to transcriptional regulation, consistent with epigenetic derepression of mitochondrial transport functions required for nucleotide biosynthesis and redox balance (Figure 10B). In contrast, SLC38A2 showed region-specific methylation heterogeneity, suggesting dynamic epigenetic control that may enable adaptive amino acid transport in response to nutrient stress (Figure 10C).
To further explore the biological consequences of these epigenetic and transcriptional states, correlations between gene expression levels and immune cell infiltration were analyzed. Expression of CP demonstrated significant associations with tumor purity and multiple immune cell populations, including B cells, CD8^+^ T cells, CD4^+^ T cells, macrophages, neutrophils, and dendritic cells, indicating a close relationship between CP-linked metabolic states and immune infiltration patterns (Figure 10D). Given CP’s role in iron metabolism and redox regulation, these associations are biologically consistent with known links between iron homeostasis, oxidative stress, and immune cell recruitment. Similarly, SLC25A13 expression correlated with tumor purity and several immune cell subsets, particularly macrophages, neutrophils, and dendritic cells (Figure 10E). As a mitochondrial aspartate–glutamate carrier, SLC25A13 supports biosynthetic and proliferative metabolism, which may indirectly shape immune infiltration by altering tumor growth dynamics and metabolic competition within the microenvironment. Expression of SLC38A2 was also significantly associated with tumor purity and immune infiltration, including CD8^+^ T cells, macrophages, neutrophils, and dendritic cells (Figure 10F). Given its function as an adaptive amino acid transporter upregulated during nutrient deprivation, SLC38A2-associated transcriptional programs likely reflect metabolic states that influence immune cell viability and function under hypoxic and nutrient-limited conditions.
Collectively, these findings demonstrate that CP, SLC25A13, and SLC38A2 are subject to gene-specific epigenetic regulation and are tightly linked to the immune landscape of GBM (Figure 10A–F). The observed associations reflect coordinated epigenetic, metabolic, and microenvironmental states rather than direct immunoregulatory roles. Nevertheless, the convergence of DNA methylation remodeling, transcriptional activation, and immune infiltration patterns supports the involvement of these genes in shaping the immunometabolic niche of GBM and provides further justification for their prioritization in downstream mechanistic and therapeutic analyses.
2.5. Single-Cell Validation of Tumor-Intrinsic Expression of CP, SLC25A13, and SLC38A2
ScRNA-seq analysis was performed to define the cellular landscape and tumor-intrinsic context of CP, SLC25A13, and SLC38A2 in GBM. Major cell populations within the tumor ecosystem were first delineated, revealing that malignant cell states dominate the cellular composition, particularly astrocyte-like and oligodendrocyte progenitor–like malignant populations, alongside smaller fractions of non-malignant oligodendrocytes and mono/macrophages (Figure 11A). Substantial inter-patient variability in lineage composition was observed, underscoring the intrinsic heterogeneity of GBM across individuals (Figure 11B). Dimensionality reduction and clustering analysis demonstrated clear transcriptional separation between malignant and non-malignant compartments, confirming robust cell-type annotation and preservation of malignant state identity (Figure 11C). Within this framework, feature plots revealed that CP, SLC25A13, and SLC38A2 expression is preferentially enriched within malignant cellular compartments rather than uniformly distributed across all cell types (Figure 11D–F). This localization supports a tumor-intrinsic role for these genes rather than expression driven by immune or stromal cell populations. Violin plot analysis further quantified expression differences across annotated cell populations, revealing cell-type–specific expression patterns (Figure 11G–I). CP expression is primarily associated with malignant subclusters linked to stress-adapted transcriptional programs, consistent with its role in iron metabolism and redox regulation. SLC25A13 expression is elevated in malignant populations characterized by high biosynthetic demand, aligning with its function in mitochondrial aspartate–glutamate exchange and nucleotide biosynthesis. SLC38A2 exhibits broad expression across malignant states, reflecting its role as an adaptive amino acid transporter supporting metabolic plasticity under nutrient-limited and hypoxic conditions. To assess how these malignant populations interact within the tumor ecosystem, intercellular communication analysis was performed. Interaction heatmaps revealed extensive communication between malignant subclusters and other tumor-associated cell types, indicating that malignant cells act as dominant signaling hubs within the GBM microenvironment (Figure 11J). Network visualization further highlighted astrocyte-like malignant cells as central interaction nodes, exhibiting strong connectivity with multiple malignant and non-malignant populations (Figure 11K). This interaction dominance suggests that malignant cells expressing CP, SLC25A13, and SLC38A2 are not only metabolically active but also structurally positioned to influence the broader tumor microenvironment through paracrine and juxtacrine signaling.
Collectively, this single-cell analysis establishes that CP, SLC25A13, and SLC38A2 are predominantly expressed within malignant GBM cell states, display cell-type–specific expression heterogeneity, and are embedded within highly interactive malignant communication networks (Figure 11A–K). These findings provide cellular-level validation of bulk transcriptomic results and support a model in which glutamine-associated metabolic genes contribute to GBM progression through tumor-intrinsic metabolic adaptation and coordinated intercellular communication.
2.6. Pharmacogenomic Drug Analysis and Structural Validation
To translate the pathway-level findings into potential therapeutic vulnerabilities, we systematically evaluated the association between CP, SLC25A13, and SLC38A2 expression and drug sensitivity profiles using large-scale pharmacogenomic datasets. Drug–gene correlation analyses were performed using the Genomics of Drug Sensitivity in Cancer (GDSC) and Cancer Therapeutics Response Portal (CTRP) resources, enabling an unbiased assessment of how transcriptional variation in these genes relates to cellular response across diverse cancer cell lines (Figure 12).
In the GDSC analysis (Figure 12A), several compounds exhibited significant negative correlations between drug response and gene expression, indicating increased drug sensitivity in cell lines with higher CP, SLC25A13, or SLC38A2 expression. Importantly, these inverse correlations were observed predominantly for drugs targeting PI3K–AKT–mTOR signaling, mitochondrial metabolism, cell-cycle regulation, and stress-response pathways, which directly aligns with the pathway enrichment patterns identified earlier by Hallmark GSEA and KEGG analyses. In contrast, although some compounds displayed stronger negative correlation magnitudes (darker blue color intensities), these agents were deprioritized when they lacked mechanistic relevance to the metabolic and signaling axes highlighted in our pathway analysis or showed inconsistent significance across datasets.
CTRP-based correlations (Figure 12B) further supported this selection strategy by demonstrating directionally consistent but more moderate associations, reinforcing the robustness of the identified drug–gene relationships while highlighting dataset-specific differences in compound coverage and screening conditions. Together, these analyses emphasize that biological concordance with pathway activation states, rather than correlation strength alone, is critical for rational drug prioritization.
Notably, although several compounds exhibited stronger correlation magnitudes in drug sensitivity analyses, they were not automatically prioritized for downstream validation. Drug selection was guided by an integrative framework that extended beyond statistical association to include biological coherence with the identified glutamine-centric metabolic programs, consistency across independent pharmacogenomic datasets, and structural compatibility with the target proteins. Compounds were excluded if they showed discordant behavior between datasets, targeted pathways unrelated to glutamine metabolism, mitochondrial function, or mTOR-linked signaling, or lacked translational feasibility for GBM therapy. Crucially, blood–brain barrier (BBB) permeability was incorporated as a decisive selection criterion, given the anatomical and pharmacological constraints of central nervous system tumors. Only compounds with documented or strongly predicted BBB penetration were advanced, ensuring that selected candidates were not only statistically and biologically relevant but also pharmacologically realistic for brain tumor treatment. Based on these integrated criteria, representative BBB-permeant compounds were subjected to structure-based molecular docking against CP, SLC25A13, and SLC38A2 (Figure 13). Structure-based molecular docking was performed to evaluate the binding feasibility and interaction stability of these pathway-aligned, pharmacogenomically selected small-molecule compounds with CP, SLC25A13, and SLC38A2 (Figure 13A–F). Docking analyses for CP demonstrated stable ligand accommodation within surface-accessible and internal cavities. Erlotinib (Figure 13A) and dasatinib (Figure 13B) exhibited favorable binding orientations supported by hydrogen bonding, hydrophobic contacts, π–π stacking, and van der Waals interactions, with predicted binding free energies of approximately −7.4 kcal/mol and −7.3 kcal/mol, respectively. These interaction profiles are consistent with CP’s proposed role in oxidative stress regulation and tumor-associated metabolic adaptation in GBM. For SLC25A13, docking revealed stable ligand positioning within transporter-associated cavities relevant to mitochondrial aspartate–glutamate exchange. AKT inhibitor VIII (Figure 13C) displayed strong binding affinity (ΔG ≈ −10.3 kcal/mol), while gefitinib (Figure 13D) showed a favorable interaction energy (ΔG ≈ −7.1 kcal/mol), supported by extensive hydrogen bonding and hydrophobic interactions. These structural features align with pathway analyses implicating SLC25A13 in anabolic metabolism, nucleotide biosynthesis, and PI3K–AKT–mTOR–associated signaling programs. Docking analyses for SLC38A2 further demonstrated ligand engagement within putative substrate or regulatory regions of this amino acid transporter. AKT inhibitor VIII (Figure 13E) and lapatinib (Figure 13F) exhibited stable binding conformations with predicted binding free energies of approximately −9.8 kcal/mol and −8.0 kcal/mol, respectively. Interaction maps highlighted hydrogen bonds, π–alkyl interactions, and hydrophobic contacts contributing to binding stability, consistent with the established role of SLC38A2 in glutamine transport and mTOR-dependent growth signaling.
Across all three targets, the observed docking energies fall within ranges typically associated with biologically meaningful protein–ligand interactions, supporting the structural plausibility of direct target engagement. Importantly, these compounds were prioritized not solely on the basis of correlation strength in drug sensitivity analyses, but through a multilayered selection strategy integrating pathway relevance, pharmacogenomic consistency, BBB permeability, and favorable molecular interaction energetics. Collectively, these findings provide structural validation for CP, SLC25A13, and SLC38A2 as therapeutically actionable metabolic nodes in GBM.
3. Discussion
GBM is characterized by extreme metabolic plasticity, enabling tumor cells to survive under hypoxic, nutrient-limited, and highly heterogeneous microenvironmental conditions [1]. Among the metabolic adaptations observed in GBM, glutamine utilization has emerged as a critical axis supporting biosynthesis, redox balance, and signaling integration. While prior studies have largely focused on enzymatic components of glutamine metabolism or well-established metabolic drivers such as IDH mutations, comparatively less attention has been given to glutamine-associated transport, exchange, and metabolic coupling nodes that regulate intracellular nutrient flux and metabolic state coordination [6,7,9]. This gap limits our understanding of how GBM selectively rewires glutamine-related programs beyond canonical pathways and restricts the identification of novel, clinically relevant biomarkers.
In this study, we employed an integrated multi-layered analytical framework to systematically dissect glutamine-associated metabolic reprogramming in GBM. Rather than assuming uniform activation of glutamine metabolism, our differential expression analyses demonstrated selective transcriptional deregulation, indicating that GBM preferentially amplifies specific transport and exchange components while suppressing others. This observation highlights an important conceptual distinction: glutamine dependency in GBM is not a global metabolic phenomenon, but rather a modular and context-dependent process driven by a subset of dominant regulatory nodes [10]. Survival analyses further refined this framework by distinguishing genes that are merely differentially expressed from those that are clinically consequential. Although IDH1 emerged as a significant survival-associated gene, it was intentionally excluded from downstream prioritization due to its extensive characterization and mutation-driven biology in glioma. Instead, we focused on CP, SLC25A13, and SLC38A2, which consistently demonstrated tumor-specific upregulation, adverse prognostic associations, and limited prior characterization in the context of GBM glutamine metabolism. This decision reflects a deliberate emphasis on novelty and translational relevance, rather than rediscovery of established metabolic paradigms [11,12,13].
Functional enrichment, pathway analyses, and protein–protein interaction networks collectively positioned CP, SLC25A13, and SLC38A2 at the intersection of metabolic stress adaptation, biosynthetic demand, and growth-associated signaling programs. CP emerged as a node linked to oxidative stress handling, iron homeostasis, and inflammatory states, all of which are central to GBM survival under hypoxic and oxidative pressure. SLC25A13, as a mitochondrial aspartate-glutamate carrier, connected mitochondrial metabolism to cytosolic nucleotide biosynthesis and redox balance, reinforcing its role in supporting rapid proliferation and DNA damage tolerance. SLC38A2, an adaptive amino acid transporter, was associated with nutrient stress responses and mTOR-linked signaling, underscoring its importance in sustaining glutamine availability in metabolically constrained tumor regions [14,15]. Importantly, protein-level validation and epigenetic analyses reinforced the biological relevance of these findings. The concordance between mRNA upregulation, protein expression, and gene-specific DNA methylation patterns suggests that CP, SLC25A13, and SLC38A2 are actively regulated components of the GBM metabolic state, rather than passive transcriptional byproducts. This multi-layer consistency strengthens their candidacy as robust biomarkers of glutamine-associated metabolic rewiring.
The integration of tumor microenvironment and immune infiltration analyses further contextualized these genes within GBM biology. Rather than implying direct immunoregulatory functions, the observed associations indicate that CP-, SLC25A13-, and SLC38A2-high tumors occupy distinct immunometabolic states, likely reflecting metabolic competition, redox imbalance, and stress signaling that shape immune cell infiltration patterns. These findings align with emerging evidence that tumor metabolism indirectly modulates immune landscapes by altering nutrient availability and metabolic byproducts [16,17]. ScRNA-seq analyses provided critical validation of tumor-intrinsic expression, demonstrating that CP, SLC25A13, and SLC38A2 are preferentially enriched within malignant cell populations rather than stromal or immune compartments. This cellular specificity confirms that the metabolic programs identified in bulk analyses are driven by cancer cells themselves, reinforcing the biological significance of these genes as intrinsic components of GBM metabolic architecture.
Finally, pharmacogenomic correlation analysis and structure-based molecular docking were incorporated as functional validation layers rather than as drug discovery endpoints. Pharmacogenomic analyses using GDSC and CTRP datasets revealed that expression levels of CP, SLC25A13, and SLC38A2 are associated with differential drug sensitivity patterns across cancer cell lines, with negative correlations indicating increased sensitivity in gene-high contexts. However, correlation magnitude alone was not used to prioritize compounds, as several drugs displaying stronger signals were excluded due to inconsistent behavior across datasets, lack of alignment with glutamine-centric pathways identified by GSEA and MetaCore analyses or limited translational relevance. Instead, representative compounds were selected based on concordance across datasets, pathway coherence, and structural feasibility. Specifically, erlotinib and dasatinib were evaluated in relation to CP [18,19,20,21], reflecting their consistent pharmacogenomic associations and compatibility with CP-associated surface and cavity regions implicated in redox regulation and stress adaptation. For SLC25A13, AKT inhibitor VIII and gefitinib were prioritized based on their alignment with PI3K–AKT–mTOR–linked anabolic and nucleotide biosynthesis pathways, as well as their stable docking conformations within transporter-associated regions relevant to mitochondrial metabolite exchange [22,23,24]. Similarly, AKT inhibitor VIII and lapatinib were examined for SLC38A2, consistent with its role in glutamine uptake, nutrient sensing, and mTORC1–MYC–driven growth signaling [25,26]. Docking analyses demonstrated stable ligand accommodation within predicted binding pockets or transporter-associated regions of CP, SLC25A13, and SLC38A2, supported by favorable binding free energies and non-covalent interaction networks, including hydrogen bonding, hydrophobic contacts, and π-interactions. Importantly, these findings were interpreted as evidence of functional plausibility rather than therapeutic efficacy, reinforcing a conservative framework in which CP, SLC25A13, and SLC38A2 are positioned as metabolically actionable nodes warranting future experimental validation rather than immediate clinical translation.
Collectively, this study advances the understanding of glutamine-associated metabolic reprogramming in GBM by shifting the focus from canonical enzymatic pathways to transport- and exchange-centric metabolic control points. CP, SLC25A13, and SLC38A2 emerge as biologically integrated, prognostically relevant, and mechanistically plausible biomarkers that capture key aspects of GBM metabolic adaptation. By bridging transcriptomics, epigenetics, pathway analysis, tumor microenvironment context, single-cell resolution, and pharmacological feasibility, our work provides a comprehensive systems-level framework for identifying and prioritizing metabolic vulnerabilities in GBM. Future studies will be required to experimentally validate the functional roles of these genes and to determine whether their modulation can effectively disrupt glutamine-dependent metabolic states in GBM. Nevertheless, the integrative strategy presented here offers a scalable blueprint for uncovering clinically relevant metabolic biomarkers and highlights the importance of transport and metabolic coupling nodes as underexplored but critical determinants of tumor behavior.
4. Materials and Methods
4.1. Study Design, Literature Review, and Gene Selection
This study employed an integration in silico framework to investigate glutamine-associated metabolic regulators in GBM using publicly available datasets and open-access bioinformatics platforms. No new biological samples were generated. The analytical workflow comprised literature-guided gene selection followed by transcriptomic profiling, survival analysis, functional enrichment, protein-level validation using immunohistochemistry data from the Human Protein Atlas, protein–protein interaction network analysis, pharmacogenomic association analysis, and structure-based molecular docking.
An initial targeted literature review was conducted using PubMed and Google Scholar to identify glutamine transporters, metabolic enzymes, and regulatory factors previously implicated in cancer biology. Reference was made to a comprehensive glutamine metabolism framework study that systematically curated key components involved in glutamine uptake, utilization, and metabolic regulation. This resource was used solely to inform unbiased gene selection, while all downstream analyses were performed independently in a GBM-specific context. Search terms included glutamine transport, SLC1A5, ASCT2, LAT1, SLC7A5, SLC6A14, glutamine metabolism, GLS, ALDH18A1, and glutamine addiction.
Based on this review, a priori gene set representing core regulators of glutamine metabolism was defined. This set included glutamine transporters (SLC1A5, SLC38A1, SLC38A2, SLC7A11, SLC25A1, SLC25A11, SLC25A12, SLC25A13, and SLC25A22), glutamine metabolism enzymes (GLS1, GLS2, GLUD1, GOT2, and GPT2), enzymes involved in hexosamine and asparagine biosynthesis (GFAT and ASNS), nucleotide synthesis enzymes participating in purine and pyrimidine pathways (PPAT, PFAS, GMPS, CPS1, CPS2, and CTPS), tricarboxylic acid cycle–related metabolic drivers and mutation-associated enzymes (IDH1 and IDH2), and signaling or growth regulators linked to metabolic control (mTORC1, MYC, mtKRAS, and mtLKB).
4.2. Differential Gene Expression and Survival Analysis
Differential gene expression analysis between GBM tumor tissue and normal brain tissue was performed using the GEPIA2 platform (http://gepia2.cancer-pku.cn/), which integrates RNA-sequencing data from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) project. Expression values were normalized as log2(TPM + 1), and tumor–normal comparisons were visualized using box plots generated by the GEPIA2 interface. Genes were considered differentially expressed based on a threshold of |log2 fold change| > 1 and a significance level of p < 0.05, following the platform’s default statistical pipeline [27].
Overall survival associations for candidate genes were evaluated using the GLIOVIS platform (http://gliovis.bioinfo.cnio.es/), which provides integrated survival analysis across multiple glioma cohorts. Patients were stratified into high- and low-expression groups based on median expression values. Kaplan–Meier survival curves were generated, and statistical significance was assessed using log-rank tests [28,29].
4.3. Clinicopathological Expression, Protein Expression Validation and Protein–Protein Interaction Network
To examine gene expression patterns across clinicopathological features, including tumor status, age, gender, and race, the UALCAN portal (http://ualcan.path.uab.edu/) was utilized. UALCAN analyzes TCGA RNA-seq data normalized as transcripts per million (TPM). Expression differences were visualized using boxplots with statistical comparisons performed using UALCAN’s default statistical framework [30].
Protein-level expression of selected genes was assessed using immunohistochemistry (IHC) data from the HPA (https://www.proteinatlas.org/). Representative IHC images from normal cerebral cortex and GBM tumor tissues were retrieved to evaluate staining intensity, localization, and cellular distribution [31].
Protein–protein interaction (PPI) networks were constructed using the STRING database (version 11.5; https://string-db.org/). High-confidence interaction networks were generated using a minimum interaction score threshold of 0.7. Both experimentally validated and predicted interactions were included to contextualize candidate genes within metabolic and signaling networks [32].
4.4. Pathway and Functional Network Enrichment Analysis
Functional and pathway analyses were performed to identify biological programs associated with gene expression in GBM using R-based workflows and curated pathway databases. Raw RNA-sequencing count data generated using the STAR workflow for the TCGA-GBM cohort were obtained as a SummarizedExperiment R package (version ≥1.36.0; Bioconductor, Buffalo, NY, USA). All computational analyses were conducted in R (version 4.4.0). Gene-level raw counts were normalized using the edgeR package (version ≥3.44.0), and expression values were subsequently transformed using the limma–voom framework (limma, version ≥3.58.0) to model mean-variance relationships inherent to count data. Samples were stratified into gene-high and gene-low groups based on the median expression level of the respective gene calculated from log2-transformed counts per million (logCPM) values. Differential expression analysis between the two groups was conducted using the limma package, and genes were ranked according to moderated t-statistics derived from the fitted linear model.
Functional enrichment analyses were performed using complementary R-based and curated approaches. GO enrichment analysis was conducted using the clusterProfiler package (version ≥4.10.0) to evaluate over-represented BP, CC, and MF associated with CP-related transcriptional programs. KEGG pathway enrichment analysis was also performed using clusterProfiler, with multiple testing correction applied using the Benjamini–Hochberg false discovery rate (FDR) method.
Gene set enrichment analysis (GSEA) was carried out using the fgsea package (version ≥1.28.0) implementing the multilevel algorithm. Ranked gene lists were generated based on limma-derived moderated t-statistics. The Hallmark gene set collection was obtained from the Molecular Signatures Database (MSigDB, version 7.5.1) using the msigdbr package (version ≥7.5.1). Enrichment significance was evaluated using permutation-based p-values followed by FDR correction, and gene sets with adjusted FDR < 0.05 were considered statistically significant [33].
Genes co-expressed with CP, SLC25A13, and SLC38A2 were identified using cBioPortal (https://www.cbioportal.org) by selecting the top 10% of genes ranked according to Pearson correlation coefficients within the TCGA-GBM cohort. These correlated gene sets were used to characterize transcriptional programs associated with high expression of each candidate gene. Functional annotation and pathway enrichment analyses were first performed using R-based approaches, followed by curated pathway validation. To complement and refine these findings, pathway enrichment analysis was further conducted using MetaCore™ (version 17.1, Clarivate Analytics, Philadelphia, PA, USA), a curated systems biology platform that integrates experimentally supported molecular interactions and manually annotated signaling networks. MetaCore analysis was used to validate and contextualize enriched pathways identified by GO and KEGG analyses, enabling prioritization of biologically relevant metabolic, signaling, and regulatory processes associated with CP, SLC25A13, and SLC38A2 [34].
4.5. DNA Methylation and Immune Infiltration Analysis and ScRNA-Seq Validation
DNA methylation profiles of candidate genes were analyzed using MethSurv (https://biit.cs.ut.ee/methsurv/, accessed on 25 February 2026), which integrates TCGA DNA methylation array data with corresponding clinical annotations. CpG-level methylation patterns were examined to assess epigenetic regulation and were correlated with gene expression levels to evaluate their potential contribution to transcriptional deregulation in GBM [35].
Tumor immune infiltration analysis was performed using TIMER 2.0 (http://timer.cistrome.org/), which estimates the abundance of immune cell populations using multiple deconvolution algorithms. Associations between gene expression and immune cell infiltration levels were evaluated with adjustment for tumor purity, allowing assessment of immune-metabolic context while minimizing confounding effects of non-tumor cells [36].
ScRNA-seq validation was conducted using the TISCH2 database (https://tisch.compbio.cn/), which provides curated and annotated single-cell transcriptomic datasets for GBM. Cell-type-specific expression patterns of candidate genes were examined using feature plots and violin plots, enabling validation of tumor-intrinsic expression and assessment of expression distribution across malignant, immune, and stromal cell populations. Cell-level expression patterns were interpreted to provide spatial and intercellular context for bulk transcriptomic findings [37].
4.6. Drug Sensitivity and Molecular Docking Analysis
Drug–gene correlation analysis was performed using the GSCA platform (https://guolab.wchscu.cn/GSCA/#/, accessed on 26 December 2025). Associations between gene expression and drug sensitivity (IC_50_) were evaluated using data from GDSC and CTRP datasets. Negative correlations indicated increased sensitivity in high-expression contexts. Statistical significance was assessed using FDR-adjusted p-values [38].
Protein structures corresponding to selected target genes were retrieved from the RCSB Protein Data Bank (https://www.rcsb.org/) or predicted using AlphaFold (https://alphafold.ebi.ac.uk/) [39], when experimental structures were unavailable. Small-molecule structures were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/) [40]. Protein and ligand structures were prepared and optimized according to standard docking protocols prior to molecular docking using AutoDock Vina (version 1.2.3; The Scripps Research Institute, La Jolla, CA, USA) [41]. Docking poses and protein–ligand interaction profiles were visualized using BIOVIA Discovery Studio 2026 (Dassault Systèmes, San Diego, CA, USA), and binding affinity scores were reported as predicted free energies of binding (kcal/mol).
4.7. Statistical Analysis
All statistical analyses were performed using R software (version 4.4.0) unless otherwise specified [42]. Continuous variables were summarized using appropriate descriptive statistics, and statistical tests were selected based on data distribution and analytical context. For transcriptomic analyses, gene expression values were log2-transformed where applicable, and differential expression analyses were conducted using established R-based frameworks as described in the corresponding Methods sections. Survival analyses were performed using the Kaplan–Meier method, with statistical significance assessed by the log-rank test. Hazard ratios (HRs) were estimated from Cox proportional hazards models where applicable, and HR values greater than 1 were interpreted as indicating an association with poorer overall survival. Patients were stratified into high- and low-expression groups based on median gene expression values unless otherwise stated. Correlation analyses between gene expression levels and other molecular or clinical variables were performed using Spearman’s rank correlation coefficient, given the non-normal distribution of transcriptomic data. For analyses involving multiple hypothesis testing, FDR correction was applied using the Benjamini–Hochberg method to control type I error. An adjusted FDR < 0.25 was considered statistically significant unless otherwise specified. Functional enrichment analyses, including GO, KEGG pathway enrichment, and GSEA, were evaluated using permutation-based or hypergeometric testing frameworks implemented in the respective software tools. Statistical significance for enrichment analyses was determined based on FDR-adjusted p-values, with thresholds defined in the corresponding Methods sections.
All statistical tests were two-sided, and a p-value < 0.05 was considered statistically significant unless otherwise indicated. Graphical representations were generated using R-based visualization packages, and all statistical interpretations were made in accordance with the assumptions and limitations of the respective analytical methods.
5. Conclusions
In conclusion, (Figure 14) this study provides a comprehensive systems-level characterization of glutamine-associated metabolic reprogramming in GBM, with a particular emphasis on transport and metabolic coupling nodes rather than canonical enzymatic pathways. Through integrative analyses spanning transcriptomics, survival stratification, functional enrichment, epigenetic profiling, tumor microenvironment context, and single-cell resolution, we identify CP, SLC25A13, and SLC38A2 as biologically coherent and clinically relevant metabolic regulators in GBM. Our findings demonstrate that GBM selectively amplifies specific glutamine-related transport and exchange mechanisms to sustain biosynthetic demand, redox balance, and growth signaling under metabolically constrained conditions. The convergence of tumor-specific expression, adverse prognostic association, coordinated pathway activation, and malignant cell-intrinsic localization underscores the importance of these genes as markers of aggressive metabolic states rather than passive correlates of tumor burden. Importantly, by deliberately excluding well-characterized metabolic drivers such as IDH1, this study highlights underexplored but functionally integrated metabolic nodes that may better capture clinically actionable aspects of GBM biology. Beyond descriptive insights, the incorporation of pharmacogenomic correlations and structural interaction analyses provides an additional layer of functional plausibility, supporting the concept that these metabolic nodes are theoretically perturbable. Although not intended as definitive therapeutic validation, this integrative framework establishes a rational foundation for future experimental and translational studies aimed at targeting glutamine-associated vulnerabilities in GBM.
Collectively, this work advances the understanding of GBM metabolism by reframing glutamine dependency as a selective, transport-driven, and context-dependent process. The identification of CP, SLC25A13, and SLC38A2 as integrated metabolic biomarkers offers new perspectives on tumor stratification and provides a scalable blueprint for uncovering metabolically actionable targets in highly heterogeneous cancers.
Limitations
Despite the strengths of this integrative multi-omics approach, several limitations should be acknowledged. First, the majority of analyses in this study are based on publicly available transcriptomic, epigenetic, and pharmacogenomic datasets. While these resources provide substantial statistical power and biological breadth, they inherently reflect population-level trends and may not fully capture patient-specific metabolic heterogeneity or temporal metabolic dynamics during disease progression and treatment. Second, although differential expressions, survival association, and pathway enrichment analyses strongly support the biological relevance of CP, SLC25A13, and SLC38A2, these findings remain correlative in nature. While single-cell analyses and protein-level validation strengthen tumor-intrinsic interpretation, direct functional experiments are required to establish causal relationships between gene modulation and metabolic or proliferative phenotypes in GBM cells. Third, the pharmacogenomic and molecular docking analyses were incorporated as supportive validation layers rather than definitive evidence of therapeutic efficacy. Drug sensitivity correlations derived from cancer cell line panels may not fully recapitulate the metabolic complexity of GBM in vivo, particularly given differences in microenvironmental constraints and blood–brain barrier dynamics. Similarly, molecular docking provides structural feasibility but does not account for dynamic protein conformations, intracellular localization, or pharmacokinetic variability. Fourth, immune infiltration and tumor microenvironment analyses were interpreted conservatively as reflective of immunometabolic states rather than direct immunoregulatory mechanisms. Additional experimental studies, including spatial transcriptomics or metabolic flux analysis in immune–tumor co-culture systems, would be required to delineate causal interactions between glutamine metabolism and immune modulation. Finally, while this study intentionally prioritized underexplored glutamine-associated metabolic nodes to enhance novelty and translational potential, it does not exclude the possibility that additional metabolic regulators outside the selected gene set contribute meaningfully to GBM biology. Future studies integrating metabolomics, isotope tracing, and patient-derived models will be essential to validate and extend the metabolic framework proposed here.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wu W. Klockow J.L. Zhang M. Lafortune F. Chang E. Jin L. Wu Y. Daldrup-Link H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance Pharmacol. Res.202117110578010.1016/j.phrs.2021.10578034302977 PMC 8384724 · doi ↗ · pubmed ↗
- 2Davis M.E. Glioblastoma: Overview of disease and treatment Clin. J. Oncol. Nurs.201620 S 210.1188/16.CJON.S 1.2-8PMC 512381127668386 · doi ↗ · pubmed ↗
- 3Aldape K. Zadeh G. Mansouri S. Reifenberger G. von Deimling A. Glioblastoma: Pathology, molecular mechanisms and markers Acta Neuropathol.201512982984810.1007/s 00401-015-1432-125943888 · doi ↗ · pubmed ↗
- 4Lassman A.B. van den Bent M.J. What Is a Glioblastoma?Oxford University Press New York, NY, USA 202310151016
- 5Panosyan E.H. Lin H.J. Koster J. Lasky J.L. In search of druggable targets for GBM amino acid metabolism BMC Cancer 20171716210.1186/s 12885-017-3148-128245795 PMC 5331648 · doi ↗ · pubmed ↗
- 6Tardito S. Oudin A. Ahmed S.U. Fack F. Keunen O. Zheng L. Miletic H. Sakariassen P.Ø. Weinstock A. Wagner A. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma Nat. Cell Biol.2015171556156810.1038/ncb 327226595383 PMC 4663685 · doi ↗ · pubmed ↗
- 7Oizel K. Chauvin C. Oliver L. Gratas C. Geraldo F. Jarry U. Scotet E. Rabe M. Alves-Guerra M.-C. Teusan R. Efficient mitochondrial glutamine targeting prevails over glioblastoma metabolic plasticity Clin. Cancer Res.2017236292630410.1158/1078-0432.CCR-16-310228720668 · doi ↗ · pubmed ↗
- 8Natarajan S.K. Venneti S. Glutamine metabolism in brain tumors Cancers 201911162810.3390/cancers 1111162831652923 PMC 6893651 · doi ↗ · pubmed ↗