Electrochemical Three‐Component Synthesis of Vinyl Sulfonamides via Decarboxylative Sulfonylation of Cinnamic Acids
Po‐Chung Chien, Harald Kelm, Georg Manolikakes

TL;DR
A new metal-free electrochemical method efficiently produces vinyl sulfonamides using cinnamic acids, SO2, and amines under mild conditions.
Contribution
A sustainable, scalable, and selective electrochemical method for synthesizing vinyl sulfonamides without metals.
Findings
The reaction uses biobased cinnamic acid derivatives and SO2 stock solutions with graphite electrodes.
It achieves high regio- and stereoselectivity under mild conditions.
Electrodes and electrolytes are reusable, supporting sustainable synthesis.
Abstract
An efficient, electrochemical three‐component reaction for the synthesis of vinyl sulfonamides from cinnamic acids, SO2, and amines is reported. This metal‐free protocol utilizes inexpensive graphite electrodes and easy‐to‐use SO2 stock solutions to facilitate a decarboxylative transformation under mild conditions. The reaction proceeds with high regio‐ and stereoselectivity. The use of cinnamic acid derivatives as biobased feedstocks, combined with the demonstrated scalability and electrode/electrolyte reusability, highlights the potential of this approach for a sustainable synthesis of the important vinyl sulfonamide scaffold. An efficient, metal‐free electrochemical three‐component reaction of cinnamic acids, SO2, and amines to access (E)‐β‐styryl sulfonamides is reported. Utilizing graphite electrodes and SO2 stock solutions, this decarboxylative protocol proceeds under mild…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
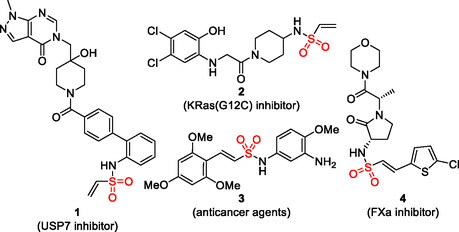 FIGURE 1
FIGURE 1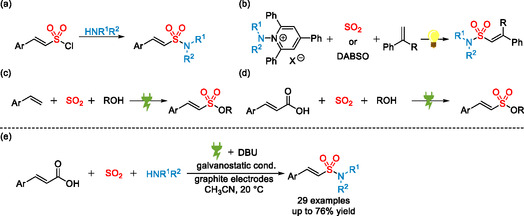 SCHEME 1
SCHEME 1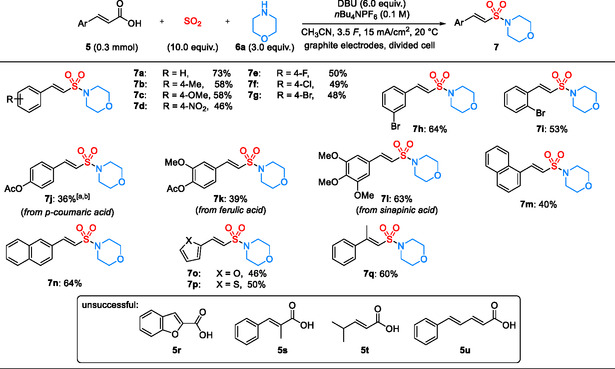 SCHEME 2
SCHEME 2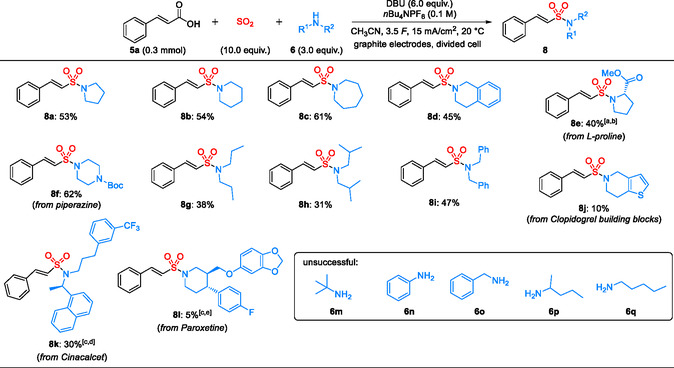 SCHEME 3
SCHEME 3 SCHEME 4
SCHEME 4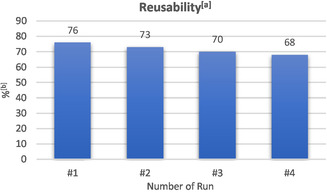 FIGURE 2
FIGURE 2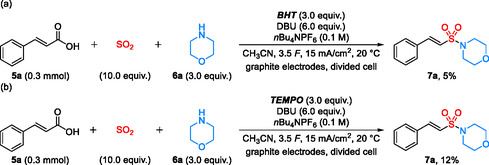 SCHEME 5
SCHEME 5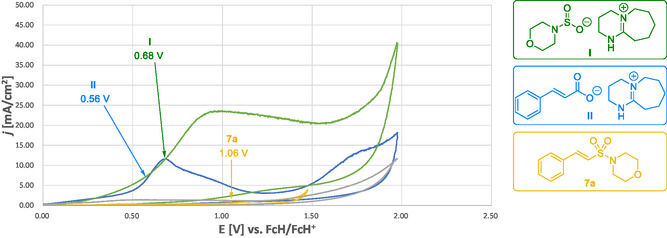 FIGURE 3
FIGURE 3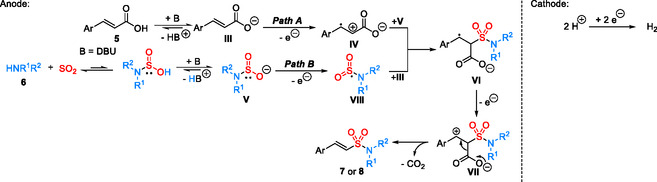 SCHEME 6
SCHEME 6|
| |||||
|---|---|---|---|---|---|
| Entry | Deviation from the standard conditions | Yield, % | Entry | Deviation from the standard conditions | Yield, % |
|
|
|
| 13 | 3.8 | 63 |
| 2 | BDD electrodes | 0 | 14 | Morpholine (2.0 equiv.) | 51 |
| 3 | Glassy carbon electrodes | 10 | 15 | Morpholine (4.0 equiv.) | 54 |
| 4 | Pt foil electrodes | 9 | 16 | DBU (4.0 equiv.) | 44 |
| 5 | Graphite anode; Pt foil cathode | 51 | 17 | DBU (8.0 equiv.) | 59 |
| 6 | 2,6‐Lutidine | 31 | 18 | SO2 (7.5 equiv.) | 56 |
| 7 | Pyridine | 13 | 19 | SO2 (12.5 equiv.) | 73 |
| 8 | DBN | 51 | 20 |
| 57 |
| 9 | DIPEA | 0 | 21 |
| 58 |
| 10 | 10 mA/cm2 | 65 | 22 |
| 56 |
| 11 | 20 mA/cm2 | 55 | 23 | No base | Traces |
| 12 | 3.0 | 60 | 24 | No electric current | 0 |
- —Bundesministerium für Forschung, Technologie und Raumfahrt
- —chungsinitiative Rheinland‐Pfalz
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSulfur-Based Synthesis Techniques · Radical Photochemical Reactions · Carbon dioxide utilization in catalysis
Introduction
1
Sulfonamides, characterized by the SO_2_–N functional group, are important compounds in medicinal and industrial chemistry [1, 2, 3, 4, 5]. As such, the development of efficient and sustainable methods for sulfonamide synthesis remains a significant goal in synthetic chemistry. One promising strategy involves the direct incorporation of sulfur dioxide (SO_2_) into organic molecules, enabling a modular and efficient synthesis of diverse sulfonamides with an increased step economy [6, 7, 8]. Potassium metabisulfite (K_2_S_2_O_5_) and DABSO (1,4‐diazabicyclo [2.2.2]octane bis(sulfur dioxide) adduct) [9, 10] have been established as convenient and easy‐to‐handle SO_2_ surrogates for this purpose [7, 8]. Recent advances also include the use of SO_2_ stock solutions, which offer a practical and accessible way to handle SO_2_ in laboratory settings [11]. As a result, efficient protocols for sulfonamide synthesis using both SO_2_ surrogates and SO_2_ stock solutions have been increasingly developed and optimized [12]. Among the various sulfonamide scaffolds, vinyl sulfonamides are of particular interest due to their electrophilic olefin moiety. Vinyl sulfonamides can be found in different covalent inhibitors, such as the Kras inhibitor or deubiquitinase USP7 inhibitor, both promising leads for cancer therapies, shown in Figure 1 [13, 14, 15]. Furthermore, vinyl sulfonamides have shown promising potential in drug discovery. In nude mouse xenograft assays, the compound (E)‐N‐(3‐amino‐4‐methoxyphenyl)‐2‐(2′, 4′, 6′‐trimethoxyphenyl)ethenesulfonamide (3) produced a marked reduction in tumor size, demonstrating its in vivo anticancer activity. In addition, GW813893 (4) has been identified as a potent, selective, and orally active factor Xa (FXa) inhibitor, providing key preclinical evidence supporting its potential as an oral antithrombotic agent (Figure 1) [16, 17, 18, 19]. Overall, vinyl sulfonamides have emerged as powerful tools in medicinal chemistry and drug development.
Representative examples of medically relevant vinyl sulfonamides.
Despite the wide‐ranging applications of vinyl sulfonamides, their synthesis remains relatively underexplored, motivating our interest in developing new synthetic strategies. Established methods for vinyl sulfonamide synthesis typically rely on vinyl sulfonyl chlorides as key intermediates (Scheme 1a) [20, 21]. The common approach for the synthesis of vinyl sulfonyl chlorides is the addition of sulfuryl chloride, a harsh reagent, to olefins [22, 23]. Therefore, this route requires an additional step for the introduction of the sulfonyl functionality and is limited due to the incompatibility of sulfuryl chloride with sensitive functional groups. In 2024, our group reported a photochemical strategy for the synthesis of vinyl sulfonamides via the direct incorporation of SO_2_ (Scheme 1b) [24, 25]. This transformation offers a complementary route involving the in situ generation of sulfamoyl radicals as key intermediates for the introduction of the sulfonamide functionality. Recently, electrochemical strategies for the synthesis of sulfonates [26, 27, 28, 29, 30, 31, 32, 33, 34], sulfonamides [32, 35, 36], and sulfamides [37] via direct SO_2_ incorporation have emerged as efficient and modular methods for assembling sulfonyl‐functionalized scaffolds in a more sustainable manner [6, 7, 38, 39]. Notably, in 2024, the Waldvogel group reported an electrochemical multicomponent synthesis of vinyl sulfonates from styrenes, alcohols, and SO_2_, enabling direct formation of the sulfonate functionality from simple starting materials (Scheme 1c) [30]. In 2025, our group disclosed an electrochemical three‐component protocol for the synthesis of vinyl sulfonates via the decarboxylation of cinnamic acids with SO_2_ incorporation (Scheme 1d) [40]. This approach enables the direct valorization of cinnamic acids, as renewable, bio‐based feedstocks, into value‐added olefin sulfonates [41, 42].
Representative approaches for the synthesis of vinyl sulfonates and vinyl sulfonamides. (a) Typical methods. (b) Photochemical sysnthesis of vinyl sulfonamides. (c) Electrochemical sysnthesis of vinyl sulfonates. (d) Electrochemical decarboxylative synthesis of vinyl sulfonates. (e) This work.
Overall, electrochemical incorporation of sulfur dioxide avoids otherwise necessary pre‐functionalization of the substrates and redox reagents, which in turn leads to a minimized amount of reagent waste, thus simplifies the work‐up, costs, and sustainability of the overall process.
Building on this strategy, we now report a so far unprecedented electrochemical three‐component reaction for the synthesis of vinyl sulfonamides, based on the decarboxylative incorporation of SO_2_ into cinnamic acids (Scheme 1e). The reaction uses simple‐to‐handle SO_2_ stock solutions and inexpensive graphite electrodes, affording β‐styryl sulfonamides directly from potentially bio‐based feedstock streams with CO_2_ and H_2_ as the only by‐products.
Results and Discussion
2
Optimization of the Reaction Conditions
2.1
Optimization studies for the synthesis of vinyl sulfonamide 7a using cinnamic acid (5a), an SO_2_ stock solution (4.6 M in MeCN, see SI for further details), and morpholine (6a) in the presence of a base, an anode electrode, and supporting electrolytes are summarized in Table 1. In the chosen electrolysis setup, SO_2_ is expected to be reduced at the cathode to form the radical anion, which can subsequently dimerize to dithionite or form complex ions with additional SO_2_ molecules [26, 35]. Therefore, a divided cell is preferred over an undivided one to prevent anodic oxidation of these species. In this divided cell setup, the catholyte contained supporting electrolytes and 5.0 equiv. (equivalents) of AcOH as a proton source for hydrogen evolution [43, 44, 45], utilizing the same electrode materials as in the anodic reaction. The best yield was obtained with inexpensive graphite electrodes, nBu_4_NPF_6_ (0.1 M) as supporting electrolyte, 1,8‐diazabicyclo [5.4.0]undec‐7‐ene (DBU) as base, a current density of 15 mA/cm^2^, and an applied charge of 3.5 F, affording the vinyl sulfonamide 7a in 73% isolated yield (Entry 1). The use of other electrodes commonly used in electrochemical SO_2_ fixation [29, 30, 31, 32, 35, 36, 37] furnished product 7a in lower yields (Entries 2–4). Common cathode materials used in electrochemical cross‐dehydrogenative coupling (CDC) reactions, such as platinum, afforded the product in lower yield (Entry 5). Other bases such as 2,6‐lutidine, pyridine, and 1,5‐diazabicyclo [4.3.0]non‐5‐ene (DBN) led to decreased yields of the sulfonamide product (Entries 6–8). No product was observed with simple amine bases, such as N,N‐diisopropylethylamine (DIPEA), presumably due to facile anodic oxidation of the amine base (Entry 9). Varying the current density (10 or 20 mA/cm^2^) or the applied amount of charge (3.0 or 3.8 F) led to decreased yields of 55–65% (Entries 10–13). Decreasing or increasing the amount of morpholine, base, SO_2_, or the concentration of supporting electrolyte resulted in reduced yields of product 7a (Entries 14–21). Replacing the supporting electrolyte nBu_4_NPF_6_ with nBu_4_NBF_4_ resulted in a lower yield (Entry 22). Only trace amounts of products were observed in the absence of a base (Entry 23). Finally, no reaction took place without applying an electric current (Entry 24). Notably, a stereoselective formation of the E‐vinyl sulfonamide was observed in all cases (E:Z > 20:1) [46].
Scope of the Reaction
2.2
Using the optimized conditions, the reaction of different cinnamic acid derivatives 5 with morpholine 6a produced the corresponding products 7a–7q in 36–73% (Scheme 2). Reactions with electron‐rich substrates afforded the corresponding vinyl sulfonamides 7b and 7c in slightly higher yields (58%) compared to reactions with electron‐poor substrates (7d or 7e; 46% and 50%). Halogenated cinnamic acids 5e–5i were well tolerated, furnishing the desired products 7e–7i in 48–64% yield, without a significant influence of the substitution pattern. Hydroxy cinnamic acids, such as ferulic acid or sinapinic acid, represent an interesting class of biobased feedstock for these three‐component transformations. In order to avoid oxidative degradation of the phenolic core [43, 44, 45], reactions with the readily accessible acetylated derivatives 5j (from p‐coumaric acid), 5k (from ferulic acid), and 5l (from sinapinic acid) were performed, furnishing the corresponding products 7j–7l in 36–63%. A reduced applied charge of 3.0 F had to be used in the synthesis of 7j to minimize product degradation under the reaction conditions. Overall, these reactions demonstrate that our process can provide a valuable approach to structurally interesting vinyl sulfonamides from hydroxycinnamic acid–based feedstocks, which are readily derived from lignocellulosic biomass [41, 42].
Scope of cinnamic acids and related substrates for the synthesis of vinyl sulfonamides 7. Isolated yields are shown. (a) The applied charge of 3.0 F was used. (b) Under the original condition, 25% of 7j was obtained.
Reactions of the two naphthyl derivatives 5m and 5n furnished the corresponding sulfonamides 7m and 7n in 40% and 64% yield. The two heterocyclic acid derivatives 5o and 5p underwent decarboxylative SO_2_ insertion, delivering the sulfonamide products 7o and 7p in 46% and 50% yield. In contrast to our previous work on vinyl sulfonate synthesis [40], no additional functionalization of the aromatic core was observed in these cases. The reaction of β‐methylcinnamic acid 5q afforded the trisubstituted vinyl sulfonamide 7q in 60% yield. Importantly, a highly stereoselective formation of the E‐configured products was observed in every case (E:Z > 20:1). Unfortunately, all attempts to convert benzofuran‐2‐carboxylic acid 5r, α‐methylcinnamic acids 5s, simple acrylic acid derivatives 5t, and conjugated dienoic acids 5u, failed under our standard reaction conditions.
Next, reactions of cinnamic acid 5a with different amines 6 were investigated using the optimized conditions (Scheme 3). Both cyclic and acyclic secondary amines were tolerated, affording the desired vinyl sulfonamides 8a–8l. Moderate to good yields of 45–61% were obtained with pyrrolidine, piperidine, azepane, and tetrahydroisoquinoline as cyclic secondary amines (compounds 8a–8d). Sulfonamide 8e was obtained in 40% yield using l‐proline methyl ester hydrochloride as the amine with an increased amount (9.0 equiv.) of DBU. Compound 8f was obtained in 62% yield using Boc‐piperazine as an amine component. Three‐component reactions with acyclic secondary amines afforded the vinyl sulfonamides 8g–8i in slightly lower yields (31–47%). Notably, the reaction of tetrahydrothieno[3,2‐c]pyridine, a building block in the synthesis of antiplatelet drug clopidogrel, afforded the product 8j in 10% yield. As before, all products were formed with excellent diastereoselectivity (E:Z > 20:1). Reactions with drugs containing structurally complex amines, such as cinacalcet and paroxetine, proceeded less efficiently, affording sulfonamides 8k and 8l in 30% and 5% yield, respectively. These reactions show that the developed process is feasible for a three‐component diversification of drug‐like or API‐based amines. The controlled installation of an electrophilic vinyl sulfonamide warhead can serve as a starting point for the efficient construction of covalent drugs from already established drug‐like scaffolds. The lower yields with tetrahydrothieno[3,2‐c]pyridine and paroxetine presumably arise from competitive oxidative degradation of the amine 6j and 6l or sulfamide formation for cinacalcet (see Supporting Information for details) [37, 43, 44, 45]. Although yields for the diversification of the selected APIs and drug‐like scaffolds are low so far, our method provides fast access to the new drug‐like scaffolds containing a covalent vinyl sulfonamide scaffold for initial biological testing. Furthermore, optimization for individual substrate combinations can lead to higher‐yielding processes.
Scope of amines for the synthesis of vinyl sulfonamides 8. Isolated yields are shown. (a) DBU (9.0 equiv.) was used. (b) Under the original condition, 16% of 8e was obtained. (c) 0.3 mmol of amine and 2.0 equiv. of cinnamic acid were used. (d) Under the original condition, 19% of 8k was obtained. (e) Under the original condition, 13% of 8l was obtained.
While secondary amines afforded the desired products, all reactions with primary amines 6m–6q were unsuccessful so far. So far, we have no conclusive explanation for the failure of primary amines. However, we assume that the formation of the key amidosulfinate intermediate is less efficient, and a competing oxidative degradation of the cinnamic acid takes place.
Scale‐Up and Reusability Tests
2.3
A 33‐fold scale‐up experiment was performed to demonstrate the applicability of this protocol (Scheme 4). The gram‐scale reaction of 5a with 6a provided vinyl sulfonamide 7a in 76% yield, with a similar result to that in the small‐scale reaction (73%; equals 43% current efficiency (CE); theoretical maximum CE (for 3.5 F) = 57%).
Scale‐up experiment.
Additionally, we evaluated the reusability of the electrolysis setup by performing the reaction under identical conditions across four consecutive runs (Figure 2). In each cycle, only the anolyte was replaced, while the catholyte and electrodes were reused without further treatment except addition of additional AcOH before each run (see Supporting Information for details). These experiments showed a slight drop in yield from 76% to 68% over four runs. Compared to our previous work on vinyl sulfonate synthesis [40], only minor electrode fouling and no deposition of a polymer layer on the anode were observed. As demonstrated by the Waldvogel group, electrolyte mixtures containing alcohol, amine, and SO_2_ can be readily recycled [36]. Recycling of both the anolyte and catholyte mixture can lead to highly sustainable processes for the synthesis of medicinally relevant vinyl sulfonamides. Moreover, the possible option to evaporate anolyte components facilitates downstream processing in large‐scale electrolysis, marking a key step toward practical implementation [47]. Together, the scale‐up and reusability tests highlight the potential for the electrification of technical organic syntheses.
Reusability tests. (a) Anolyte: 5a (0.3 mmol, 0.1 M), SO2 (10.0 equiv.), 6a (3.0 equiv.), DBU (6.0 equiv.), nBu4NPF6 (0.1 M), CH3CN, divided cell (glass frit), graphite electrodes, 15 mA/cm2, 3.5 F, 20°C. Catholyte: nBu4NPF6 (0.1 M), AcOH (5.0 equiv.), CH3CN. (b) 1H NMR yield with the use of CHPh3 as the internal standard.
Mechanistic Studies
2.4
Control experiments and cyclic voltammetry studies were conducted to elucidate the reaction mechanism. The addition of radical scavengers such as 2,6‐di‐tert‐butyl‐4‐methylphenol (BHT) (Scheme 5a) or 2,2,6,6‐tetramethylpiperidinyl‐oxyl (TEMPO) (Scheme 5b) significantly suppressed product formation as well as conversion of the cinnamic acid 5a. However, no direct trapping products derived from the radical scavengers were observed.
Control experiments.
Cyclic voltammetry studies show that under standard conditions (excess base), the cinnamate salt II undergoes oxidation at a half‐wave potential of 0.56 V before the amidosulfinate I or overoxidation of the vinyl sulfonamide 7a (Figure 3; see Supporting Information Figures S8–S12 for cyclic voltammograms for all individual components and mixtures).
Cyclic voltammograms: cinnamate II (blue), amidosulfinate intermediate I (green), vinyl sulfonamide 7a (yellow), and blank measurement (0.1 M nBu4NPF6 in MeCN; grey). The numbers given refer to the half‐wave oxidation potential of the respective compounds or mixtures.
Based on these results and literature precedents [48, 49, 50, 51, 52], we propose that a pseudo‐Kolbe‐type reaction pathway involving oxidation of the carbon scaffold is operative, rather than a classical Kolbe‐type direct oxidation of the carboxylic acid (Scheme 6). Initially, the cinnamic acid is deprotonated to form the corresponding carboxylate III. Subsequent one‐electron oxidation of III generates radical cation IV. The amidosulfinate species V is formed in situ from amine 6 and SO_2_, with DBU facilitating the equilibrium shift toward the deprotonated form [35, 36, 37]. Regioselective addition of V to IV yields the stabilized benzylic radical VI (Path A). A second anodic oxidation of VI produces the benzylic cation VII, which undergoes decarboxylation to afford the final products 7 and 8. Although the reaction mechanism can be rationalized thermodynamically based on the oxidation potentials obtained from cyclic voltammetry, the kinetic perspective should also be considered (Path B). Specifically, the amidosulfinate species V is oxidized first to generate the S‐centered radical intermediate VIII, preceding oxidation of the carboxylate III. Regioselective addition of VIII to III then affords the intermediate VI, which undergoes subsequent oxidation and decarboxylation to yield the final products. The stereoselective formation of the E‐isomer likely proceeds via the most stable staggered conformation under kinetic control. On the cathodic side, hydrogen evolution from acetic acid serves as a simple and efficient counter‐reaction.
Proposed reaction pathway.
Conclusion
3
In summary, an electrochemical three‐component reaction for the synthesis of vinyl sulfonamides from cinnamic acids, amines, and a SO_2_ stock solution, using inexpensive graphite electrodes, is described. The method demonstrates a broad substrate scope (29 examples, up to 76% yield) and is compatible with structurally complex amines, enabling a late‐stage functionalization of drug‐like molecules. Mechanistic studies provide evidence for a pseudo‐Kolbe‐type decarboxylation pathway, enabling regioselective access to vinyl sulfonamides. Hence, this method extends the scope of still less‐explored pseudo‐Kolbe‐type decarboxylations to novel scaffolds and functionalities. The potential applicability of this process in technical organic syntheses was demonstrated with scale‐up experiments and electrode/electrolyte reusability studies. Furthermore, the direct conversion of biomass‐derived cinnamic acid opens an intriguing approach for the sustainable synthesis of vinyl sulfonamides for medical or materials applications. Studies to expand the applicability of decarboxylative SO_2_‐insertion reactions to other scaffolds are currently ongoing in our laboratories.
Notes
An initial version of the manuscript was deposited to the ChemRxiv repository prior to submission [53].
Supporting Information
Additional supporting information can be found online in the Supporting Information section. Supporting Information File 1: Experimental details, spectral and crystal data, DOIS, and copies of NMR spectra for all compounds prepared in this study. X‐Ray Data: cif and checkcif files for compound 7a (CCDC 2472612). The authors have cited additional references within the Supporting Information [55, 56, 57]. Supporting Fig. S1: IKA ElectraSyn 2.0 and IKA Carousell. Supporting Fig. S2: IKA Pro‐Divide divided cell. Supporting Fig. S3: Rohde & Schwarz HMP4040 power source. Supporting Fig. S4: Electrochemical glass cell used for the scale‐up experiment equipped with a P4 frit, graphite electrodes, caps, silicone septa, and electrode holders. Scale is given in cm. Supporting Fig. S5: Scale‐up experiment in a 2 x 100 mL glass cell before (left) and after (right) the electrolysis. Supporting Fig. S6: Tests for reusing the electrodes, glass frit membrane, and catholyte. ^[a]^Conditions described above. ^[b]1^H‐NMR yield with the use of CHPh_3_ as the internal standard. Supporting Fig. S7: Unsuccessful derivatives under the standard conditions. Supporting Fig. S8: Cyclic voltammograms of cinnamate II (blue), cinnamic acid 5a (blue), morpholine 6a (dark blue), amidosulfinate intermediate I (green), DBU (brown), vinyl sulfonamide 7a (yellow), and blank measurement (0.1 M nBu_4_NPF_6_ in CH_3_CN; grey). Supporting Fig. S9: Cyclic voltammograms of cinnamate II (blue), cinnamic acid 5a (blue), DBU (brown), and blank measurement (0.1 M nBu_4_NPF_6_ in CH_3_CN; grey). Supporting Fig. S10: Cyclic voltammograms of morpholine 6a (dark blue), amidosulfinate intermediate I (green), DBU (brown), and blank measurement (0.1 M nBu_4_NPF_6_ in CH_3_CN; grey). Supporting Fig. S11: Cyclic voltammograms of cinnamate II. Supporting Fig. S12: Cyclic voltammograms of (E)‐4‐(styrylsulfonyl)morpholine 7a. Supporting Fig. S13: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7a. Supporting Fig. S14: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7b. Supporting Fig. S15: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7c. Supporting Fig. S16: ^1^H (DMSO‐d 6, 400 MHz) and ^13^C{^1^H} (DMSO‐d 6, 101 MHz) NMR Spectrum of 7d. Supporting Fig. S17: ^1^H (CDCl_3_, 400 MHz), ^13^C{^1^H} (CDCl_3_, 101 MHz), and ^19^F{^1^H} (CDCl_3_, 376 MHz) NMR Spectrum of 7e. Supporting Fig. S18: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7f. Supporting Fig. S19: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7g. Supporting Fig. S20: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7h. Supporting Fig. S21: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7i. Supporting Fig. S22: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7j. Supporting Fig. S23: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7k. Supporting Fig. S24: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7l. Supporting Fig. S25: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7m. Supporting Fig. S26: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7n. Supporting Fig. S27: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7o. Supporting Fig. S28: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7p. Supporting Fig. S29: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 7q. Supporting Fig. S30: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8a. Supporting Fig. S31: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8b. Supporting Fig. S32: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8c. Supporting Fig. S33: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8d. Supporting Fig. S34: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8e. Supporting Fig. S35: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8f. Supporting Fig. S36: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8g. Supporting Fig. S37: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8h. Supporting Fig. S38: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8i. Supporting Fig. S39: ^1^H (CDCl_3_, 400 MHz) and ^13^C{^1^H} (CDCl_3_, 101 MHz) NMR Spectrum of 8j. Supporting Fig. S40: ^1^H (CDCl_3_, 400 MHz), ^13^C{^1^H} (CDCl_3_, 101 MHz), and 19^F^{^1^H} (CDCl^3^, 376 MHz) NMR Spectrum of 8k. Supporting Fig. S41: ^1^H (CDCl_3_, 400 MHz), ^13^C{^1^H} (CDCl_3_, 101 MHz), and ^19^ ^F^{^1^H} (CDCl_3_, 376 MHz) NMR Spectrum of 9k. Supporting Fig. S42: ^1^H (CDCl_3_, 400 MHz), ^13^C{^1^H} (CDCl_3_, 101 MHz), and ^19F^{^1^H} (CDCl_3_, 376 MHz) NMR Spectrum of 8l. Supporting Table S1: Screening of electrodes, applied charge, current density, base, electrolyte, and solvent. Supporting Table S2: Screening of the stoichiometry of morpholine, DBU, SO_2_, and other parameters. Supporting Table S3: Control experiments. Supporting Table S4: Crystal data for 7a (CCDC no. 2472612, the thermal ellipsoid drawn at 50% probability level).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1H. Bisharat , Z. Ahmad , I. Fatima Ahsan , Z. Haq , T. Azam , and F. Ahmad , “The Evolving Role of Sulfonamides in Medicine and Drug Development: A Brief Review,” Advanced Journal of Chemistry, Section B: Natural Products and Medical Chemistry 7 (2025): 130.
- 2W. Liu , J. Chen , and W. Su , “Recent Advances in the Synthesis of Sulfonamides Intermediates,” Pharmaceutical Fronts 06 (2024): e 355.
- 3P. de Groote , J. Devaux , and P. Godard , “Effect of Benzenesulfonamide Plasticizers on the Glass-Transition Temperature of Semicrystalline Polydodecamide,” Journal of Polymer Science Part B 40 (2002): 2208.
- 4H. E. Gaffer , M. R. Elgohary , H. A. Etman , and S. Shaaban , “Antibacterial Evaluation of Cotton Fabrics by using Novel Sulfonamide Reactive Dyes,” Pigment & Resin Technology 46 (2017): 210.
- 5S. I. Kang and Y. H. Bae , “p H-Induced Solubility Transition of Sulfonamide-Based Polymers,” Journal of Controlled Release 80 (2002): 145.11943394 10.1016/s 0168-3659(02)00021-4 · doi ↗ · pubmed ↗
- 6P. Vogel , M. Turks , L. Bouchez , D. Marković , A. Varela‐Álvarez , and J. Á. Sordo , “New Organic Chemistry of Sulfur Dioxide,” Accounts of Chemical Research 40 (2007): 931.17685582 10.1021/ar 700096 h · doi ↗ · pubmed ↗
- 7G. Liu , C. Fan , and J. Wu , “Fixation of Sulfur Dioxide into Small Molecules,” Organic & Biomolecular Chemistry 13 (2015): 1592.25502340 10.1039/c 4ob 02139 h · doi ↗ · pubmed ↗
- 8K. Hofman , N.‐W. Liu , and G. Manolikakes , “Radicals and Sulfur Dioxide: A Versatile Combination for the Construction of Sulfonyl-Containing Molecules,” Chemistry: A European Journal 24 (2018): 11852.29315874 10.1002/chem.201705470 · doi ↗ · pubmed ↗