Genome resources—A chromosome-level genome assembly for the long-nosed leopard lizard, Gambelia wislizenii, the first reference genome for the lizard family Crotaphytidae
Jimmy A McGuire, Jonathan Q Richmond, Merly Escalona, Mohan P A Marimuthu, Oanh Nguyen, Samuel Sacco, Eric Beraut, Erin Toffelmier, Robert D Cooper, Michael Westphal, Robert N Fisher, Ian J Wang, H Bradley Shaffer

TL;DR
This paper presents the first chromosome-level genome assembly for the long-nosed leopard lizard, a key resource for studying genetic diversity and traits in this lizard family.
Contribution
The study provides the first reference genome for the lizard family Crotaphytidae, using advanced sequencing technologies and annotation methods.
Findings
The genome assembly has a total length of ~2.47 Gb with a scaffold N50 of 380.1 Mb and 97.4% BUSCO completeness.
Transcriptome-based annotation identified 23,279 genes with 98.9% BUSCO completeness.
The genome will support research on hybridization dynamics and genetic traits in Gambelia species.
Abstract
We report on an annotated chromosome-level genome assembly for the long-nosed leopard lizard, Gambelia wislizenii, as part of the California Conservation Genomics Project (CCGP). All 17 species of reptiles, including two turtles, seven lizards, and seven snakes targeted for reference genome sequencing by the CCGP are now complete and posted on NCBI, and this article is the third of seven CCGP lizard release papers to be published. It is also the first species of the family Crotaphytidae to have a released reference genome. Following the CCGP pipeline, the G. wislizenii genome was produced using Pacific Biosciences HiFi long reads and Omni-C proximity ligation data. The de novo assembly includes 69 scaffolds and has a total length of ~ 2.47 Gb, a scaffold N50 length of 380.1 Mb, and a BUSCO completeness score of 97.4% based on the tetrapod gene set. We improved the annotation of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Fig. 1
Fig. 1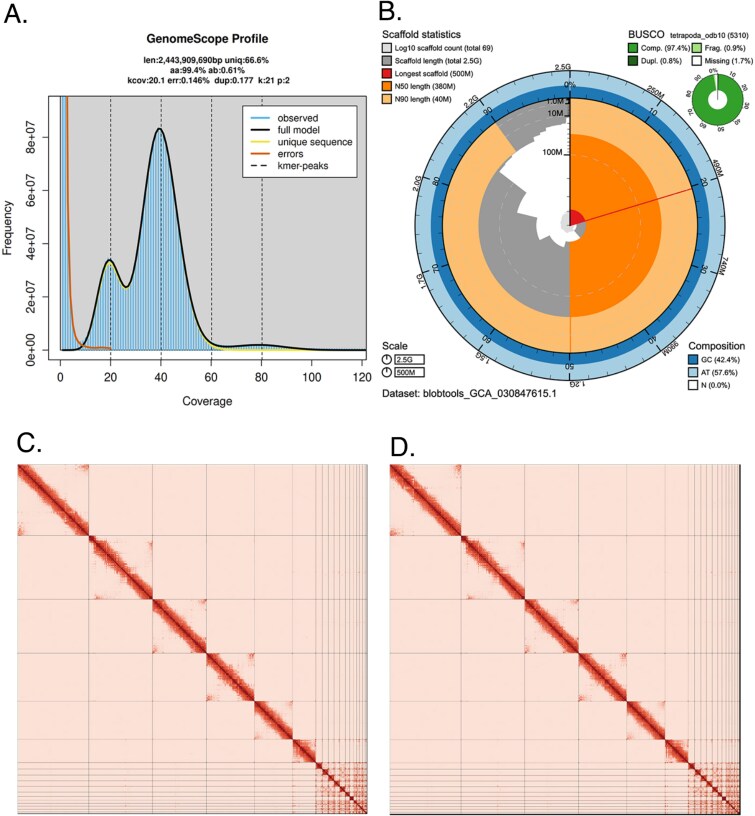 Fig. 2
Fig. 2|
|
|
|
|---|---|---|
|
| ||
| Filtering PacBio HiFi adapters | HiFiAdapterFilt | Commit 64d1c7b |
| K-mer counting | Meryl ( | 1 |
| Estimation of genome size and heterozygosity | GenomeScope | 2 |
| De novo assembly (contiging) | HiFiasm (Hi-C Mode, -primary, output hic.hap1.p_ctg, hic.hap2.p_ctg) | 0.16.1-r375 |
|
| ||
| Omni-C data alignment | Arima Genomics Mapping Pipeline | Commit 2e74ea4 |
| Arima Genomics Mapping Pipeline (AGMP) | BWA-MEM | 0.7.17-r1188 |
| samtools | 1.11 | |
| filter_five_end.pl (AGMP) | Commit 2e74ea4 | |
| two_read_bam_combiner.pl ((AGMP)) | Commit 2e74ea4 | |
| picard | 2.27.5 | |
| Omni-C scaffolding | SALSA (-DNASE, -i 20, -p yes) | 2 |
|
| ||
| Short-read alignment | BWA-MEM (-5SP) | 0.7.17-r1188 |
| SAM/BAM processing | samtools | 1.11 |
| SAM/BAM filtering | pairtools | 0.3.0 |
| Pairs indexing | pairix | 0.3.7 |
| Matrix generation | cooler | 0.8.10 |
| Matrix balancing | hicExplorer (hicCorrectmatrix correct --filterThreshold -2 4) | 3.6 |
| Contact map visualization | HiGlass | 2.1.11 |
| PretextMap | 0.1.4 | |
| PretextView | 0.1.5 | |
| PretextSnapshot | 0.0.3 | |
| Manual curation tools | Rapid curation pipeline (Wellcome Trust Sanger Institute, Genome Reference Informatics Team) | Commit 7acf220c |
|
| ||
| Basic assembly metrics | QUAST (--est-ref-size) | 5.0.2 |
| Assembly completeness | BUSCO (-m geno, -l tetrapoda) | 5.0.0 |
| Merqury | 29 January 2020 | |
|
| ||
| Local alignment tool | BLAST+ (-db nt, -outfmt ‘6 qseqid staxids bitscore std’, -max_target_seqs 1, -max_hsps 1, -evalue 1e-25) | 2.15 |
| General contamination screening | BlobToolKit (HiFi coverage, BUSCO = tetrapoda, NCBI Taxa ID = 43593) | 2.3.3 |
|
| ||
| Mitochondrial genome assembly | MitoHiFi (-r, -p 90, -o 1) Reference: | 2.2 |
| MitoHiFi: Mitochondrial genome annotation | MitoFinder (MitoHiFi pipeline parameters) | 1.4 |
|
| ||
| Genome annotation | NCBI Eukaryotic Genome Annotation Pipeline (“ | 0.3.2-alpha |
| Local alignment of transcripts and proteins | BLAST | |
| HMM-based gene prediction | NCBI Gnommon pipeline | |
| Alingment of RNA-seq reads | STAR | |
| Gene identification | BUSCO | |
| Bio projects and vouchers | CCGP NCBI BioProject | PRJNA720569 | |||||
| Genus NCBI BioProject | PRJNA765827 | ||||||
| Species NCBI BioProject | PRJNA986198 | ||||||
| Transcriptome NCBI BioProject | PRJNA1011925 | ||||||
| NCBI Assembly BioSample | SAMN35845454 | ||||||
| NCBI Transcriptome BioSamples | SAMN40863703, SAMN40863704, SAMN40863705, SAMN40863706, SAMN40863707, SAMN40863708, SAMN40863709 | ||||||
| Specimen identification | HBS 135999 | ||||||
| NCBI Genome accessions |
|
| |||||
| Assembly accession | |||||||
| Genome sequences | GCA_030847625.1 | GCA_030847615.1 | |||||
| Data Dryad Annotation accession | TBD | ||||||
| Genome sequence | PacBio HiFi reads | Run | 1 PACBIO_SMRT (Sequel IIe) run: 7.8 M spots, 101.2G bases, 59.6Gb | ||||
| Accession | SRX25151365 | ||||||
| Omni-C Illumina reads | Run | 2 ILLUMINA (Illumina NovaSeq 6000) runs: 496.4 M spots, 149.9G bases, 49.5Gb | |||||
| Accession | SRX25151366–7 | ||||||
| Genome assembly quality metrics | Assembly identifier (Quality code | rGamWis1(8.8.P8.Q68.C99) | |||||
| HiFi Read coverage | 41.41X | ||||||
|
|
| ||||||
| Number of contigs | 169 | 96 | |||||
| Contig N50 (bp) | 141,377,706 | 138,191,064 | |||||
| Contig NG50 | 141,377,706 | 138,191,064 | |||||
| Longest Contigs | 293,118,657 | 321,148,149 | |||||
| Number of scaffolds | 141 | 69 | |||||
| Scaffold N50 | 379,351,600 | 380,060,318 | |||||
| Scaffold NG50 | 379,351,600 | 380,060,318 | |||||
| Largest scaffold | 501,950,513 | 502,046,835 | |||||
| Size of final assembly | 2,469,046,425 | 2,466,589,883 | |||||
| Phased block NG50 | 141,377,706 | 138,191,064 | |||||
| Gaps per Gbp (# Gaps) | 11(28) | 11(27) | |||||
| Indel QV (Frame shift) | 48.49 | 48.58 | |||||
| Base pair QV | 67.803 | 68.4573 | |||||
| Full assembly = 68.1177 | |||||||
| k-mer completeness | 89.2806 | 89.2926 | |||||
| Full assembly = 99.5467 | |||||||
| BUSCO completeness |
|
|
|
|
| ||
| H1 | 97.40% | 96.60% | 0.80% | 0.80% | 1.80% | ||
| H2 | 97.40% | 96.60% | 0.80% | 0.90% | 1.70% | ||
| Organelles | # Partial mitochondrial sequence | ||||||
| Genome annotation quality metrics |
| ||||||
| Genes | 23,279 | ||||||
| Transcripts | 53,856 | ||||||
| mRNA | 48,567 | ||||||
| lncRNA | 5058 | ||||||
| CDSs | 48,579 | ||||||
| BUSCO completeness |
|
|
|
|
| ||
| (eukaryota_odb12) | 99.20% | 99.20% | 0.00% | 0.00% | 0.80% | ||
| (tetrapoda_odb10) | 98.80% | 98.00% | 0.80% | 0.40% | 0.70% | ||
| (squamata_odb12) | 98.80% | 98.40% | 0.40% | 0.50% | 0.80% | ||
- —NIH10.13039/100000002
- —National Institutes of Health10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmphibian and Reptile Biology · Turtle Biology and Conservation · Genomics and Phylogenetic Studies
Leopard lizards of the genus Gambelia are widespread denizens of arid landscapes across much of western North America. These relatively large lizards are well-known for their predatory habits, preying on a diversity of small vertebrates and arthropods (Tanner and Krogh 1974; Parker and Pianka 1976; Tollestrup 1983). The genus contains three species, all of which occur in California. The long-nosed leopard lizard, Gambelia wislizenii, is the most broadly distributed, with a range including Sonoran, Mohave, Great Basin, and Chihuahuan desert habitats. In contrast, the state and federally endangered blunt-nose leopard lizard, Gambelia sila, is endemic to California’s San Joaquin Desert (see Germano et al. 2011; Stewart et al. 2019), and Cope’s leopard lizard, Gambelia copei, is found throughout much of the Baja California Peninsula, barely entering California at a few documented sites adjacent to the Mexican border (McGuire 1996; Mahrdt et al. 2010). Because a primary aim of the California Conservation Genomics Project (CCGP) is to document landscape-scale genetic diversity across the state for a diverse set of taxa (Fiedler et al. 2022; Shaffer et al. 2022), a high-quality reference genome for any one species can be paired with re-sequenced genomes for each of the three Gambelia species, providing landscape genomic data spanning most of the arid landscapes of the state.
The three Gambelia species share numerous phenotypic and ecological traits. All three species are relatively large and share the unusual habit (for a lizard) of consuming vertebrates, especially other lizards, as part of their diets (although G. sila is thought to prey on vertebrates less frequently than the larger G. wislizenii and G. copei; Montanucci 1965; Germano et al. 1994, 2007). Like other crotaphytids, gravid females of all three species display conspicuous red or orange coloration that develops shortly before ovulation and dissipates post-parturition. However, Gambelia species also exhibit important interspecific differences. In particular, G. wislizenii and G. copei exhibit female-biased sexual size dimorphism apparently tied to their loss of territoriality, whereas the territorial G. sila exhibits male-biased size dimorphism that is typically associated with territorial defense (McCoy 1967; Montanucci 1970; McGuire 1996). Perhaps as a result, the non-territorial G. copei and G. wislizenii lack male breeding coloration, whereas G. sila males exhibit vibrant pink to rusty red coloration on the head and trunk during the breeding period (Montanucci 1965). Gambelia wislizenii and G. copei also differ from G. sila in having relatively longer snouts than the aptly named blunt-nosed leopard lizard (McGuire 1996; Lappin and Swinney 1999). All three species are closely related and were previously considered to be conspecific, and gene flow has been documented between G. wislizenii and G. sila (Montanucci 1970; Richmond et al. 2017) and may occur between G. wislizenii and G. copei where they meet in Paseo de San Matias in northern Baja California, Mexico (McGuire 1996).
Here, we present the first annotated assembled reference genome for G. wislizenii, generated as part of the CCGP (Beninde et al. 2022; Fiedler et al. 2022; Shaffer et al. 2022; Toffelmier et al. 2022). The G. wislizenii genome assembly joins 39 other genomes representing the cosmopolitan iguanian lizard clade, including two other CCGP species: Blainville’s horned lizard, Phrynosoma blainvillii (Richmond et al. 2023), and the western fence lizard, Sceloporus occidentalis (Bishop et al. 2023). It is the first reference genome for the iguanian family Crotaphytidae. Combined with the landscape genomics approach of the CCGP, this genome will be a powerful tool enabling researchers to characterize hybridization dynamics between G. wislizenii and G. sila and possibly G. wislizenii and G. copei, document the remaining diversity within G. sila relative to its more widespread congeners, and explore the genetic underpinnings of key traits that vary between the three Gambelia species.
Methods
Biological materials
We captured, euthanized, and flash froze multiple tissues (blood, brain, intestine, liver, lung, muscle, and ovary) from a G. wislizenii collected from the mouth of French Canyon, Inyo Mountains, Inyo County, California (Fig. 1: 36.6565016, −118.0111543) on 23 April 2021 (CDFW entity permit no. SC-838). The specimen (female, field number HBS 135999, MVZ:Herp:300824) was euthanized, tissues were harvested, and the voucher was preserved in formalin and accessioned at the Museum of Vertebrate Zoology, University of California at Berkeley for further research use.
Distribution of Gambelia wislizenii (blue), G. sila (orange), and G. copei (magenta) in California modified from Hansen and Shedd (2025). The yellow dot indicates the collection site for the genome voucher specimen (mouth of French Canyon, Inyo Mountains, Inyo County (36.6565016, −118.0111543). A) Adult male G. wsislizenii basking in San Juan Co., Utah (B. Oldfield); B) gravid female G. sila from the Carrizo Plain National Monument, San Luis Obispo County illustrating gravid coloration (B. Bouton, CC BY-NC-SA); C) G. copei from Campo, San Diego County (JQ Richmond); and D) G. wislizenii reference genome voucher sample from the mouth of French Canyon, Inyo Mountains, Inyo County (H.B. Shaffer).
High-molecular-weight genomic DNA isolation
We extracted high molecular weight (HMW) genomic DNA (gDNA) from 30 mg of liver tissue using the Nanobind Tissue Big DNA kit as per the manufacturer’s instructions (Pacific BioSciences—PacBio, Menlo Park, CA). We estimated DNA purity based on absorbance ratios (260/280 = 1.83 and 260/230 = 2.11) from a NanoDrop ND-1000 spectrophotometer and quantified the final DNA yield (11.2 μg) using Qubit 2.0 Fluorometer (Thermo Fisher Scientific, MA). Eighty-eight percent of the DNA fragments were 50 kb or longer based on the sizing of HMW gDNA on the Femto Pulse system (Agilent, Santa Clara, CA).
HiFi library preparation and sequencing
The HiFi SMRTbell library was constructed using the SMRTbell Express Template Prep Kit v2.0 (PacBio, Cat. #100-938-900) according to the manufacturer’s instructions. HMW gDNA was sheared to a target DNA size distribution between 15 and 18 kb using Diagenode’s Megaruptor 3 system (Diagenode, Belgium; Cat. B06010003). The sheared gDNA was concentrated using 0.45X of AMPure PB beads (PacBio, Cat. #100-265-900) for the removal of single-strand overhangs at 37°C for 15 min. This was followed by enzymatic DNA damage repair at 37°C for 30 min, end repair and A-tailing at 20°C for 10 min and 65°C for 30 min, and the ligation of overhang adapters v3 at 20°C for 60 min. The SMRTbell library was purified and concentrated with 1X AMPure PB beads for nuclease treatment at 37°C for 30 min and size selection using the PippinHT system (Sage Science, Beverly, MA; Cat #HPE7510) to collect fragments greater than 7 to 9 kb. The 15 to 20 kb average HiFi SMRTbell library was sequenced at UC Davis DNA Technologies Core (Davis, CA) using four 8 M SMRT cells, Sequel II sequencing chemistry 2.0, and 30-h movies each on a PacBio Sequel II sequencer.
Omni-C library preparation
The Omni-C library was prepared using the Dovetail Omni-C Kit (Dovetail Genomics, CA) according to the manufacturer’s protocol with slight modifications. First, specimen tissue (liver, ID: HBS 135999) was thoroughly ground with a mortar and pestle while cooled with liquid nitrogen. Subsequently, chromatin was fixed in place in the nucleus. The suspended chromatin solution was then passed through 100 and 40 μm cell strainers to remove large debris. Fixed chromatin was digested under various conditions of DNase I until a suitable fragment length distribution of DNA molecules was obtained. Chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter containing ends. After proximity ligation, crosslinks were reversed and the DNA purified from proteins. Purified DNA was treated to remove biotin that was not internal to ligated fragments. An NGS library was generated using an NEB Ultra II DNA Library Prep Kit (New England Biolabs, Ipswich, MA) with an Illumina compatible y-adaptor. Biotin-containing fragments were then captured using streptavidin beads. The post capture product was split into two replicates prior to polymerase chain reaction enrichment to preserve library complexity with each replicate receiving unique dual indices. The library was sequenced at Vincent J. Coates Genomics Sequencing Lab (Berkeley, CA) on an Illumina NovaSeq platform (Illumina, CA) to generate approximately 100 million 2 × 150 bp read pairs per GB genome size.
Transcriptome RNA extraction, library preparation, and sequencing
Total RNA was extracted from seven separate tissues from the same individual (blood, brain, intestine, liver, lung, muscle, and ovary) using a Qiagen RNeasy Mini Kit (Qiagen, Netherlands) according to the manufacturer’s protocol. RNA libraries were then prepared using the KAPA mRNA HyperPrep Kit (Roche, Switzerland) according to the manufacturer’s protocol. Libraries were sequenced using 150 bp paired end reads on an Illumina NovaSeq 6000 platform (Illumina, San Diego, CA), to generate approximately 50 M reads per library.
Nuclear genome assembly
We assembled the genome of a female G. wislizenii following the CCGP assembly pipeline Version 5.0, as outlined in Table 1 where we list the tools and non-default parameters used in the assembly process. We removed the remnant adapter sequences from the PacBio HiFi dataset using HiFiAdapterFilt (Sim et al. 2022) and generated an initial diploid phased assembly using HiFiasm (Cheng et al. 2022) in “HiC” mode with the filtered PacBio HiFi reads and the Omni-C short-reads. This generates two assemblies, one per haplotype. We then aligned the Omni-C data to both assemblies separately following the Arima Genomics Mapping Pipeline (https://github.com/ArimaGenomics/mapping_pipeline) and then scaffolded both assemblies with SALSA (Ghurye et al. 2017; Ghurye et al. 2019).
The assemblies for both haplotypes were manually curated by iteratively generating and analyzing their corresponding Omni-C contact maps. To generate the contact maps we aligned the Omni-C data with BWA-MEM (Li 2013), identified ligation junctions, and generated Omni-C pairs (Lee et al. 2022) using pairtools (Open2C et al. 2024). Then, we generated multi-resolution Omni-C matrices with cooler (Abdennur and Mirny 2020) and balanced them with hicExplorer (Ramírez et al. 2018). We used HiGlass (Kerpedjiev et al. 2018) and the PretextSuite (https://github.com/wtsi-hpag/PretextView; https://github.com/wtsi-hpag/PretextMap; https://github.com/wtsi-hpag/PretextSnapshot) to visualize the contact maps. We identified misassemblies and misjoins in these contact maps and modified the assemblies using the Rapid Curation pipeline from the Wellcome Trust Sanger Institute, Genome Reference Informatics Team (https://gitlab.com/wtsi-grit/rapid-curation). Some of the remaining gaps (joins generated during scaffolding and/or curation) were closed using the PacBio HiFi reads and YAGCloser (https://github.com/merlyescalona/yagcloser). We checked for contamination using the BlobToolKit Framework (Challis et al. 2020).
Mitochondrial genome assembly
We assembled the mitochondrial genome from the PacBio HiFi reads using the reference-guided pipeline MitoHiFi (Allio et al. 2020; Uliano-Silva et al. 2023). The mitochondrial sequence of a G. wislizenii (NCBI:NC_012831.1) was used as the starting sequence. After completion of the nuclear genome, we searched for matches of the resulting mitochondrial assembly sequence in the nuclear genome assembly using BLAST+ (Camacho et al. 2009) and filtered out contigs and scaffolds from the nuclear genome with a percentage of sequence identity > 99% and size smaller than the mitochondrial assembly sequence.
Genome size estimation and quality assembly assessment
We generated k-mer counts from the PacBio HiFi reads using meryl (https://github.com/marbl/meryl). The k-mer counts were then used in GenomeScope2.0 (Ranallo-Benavidez et al. 2020) to estimate genome features including genome size, heterozygosity, and repeat content. To obtain general contiguity metrics, we ran QUAST (Gurevich et al. 2013). To evaluate genome quality and functional completeness, we used BUSCO (Manni et al. 2021) with the Tetrapoda ortholog database (tetrapoda_odb10) which contains 5,286 genes. Assessment of base level accuracy (QV) and k-mer completeness was performed using the previously generated meryl database and merqury (Rhie et al. 2020). We further estimated genome assembly accuracy via BUSCO gene set frameshift analysis using the pipeline described by Korlach et al. (2017). Measurements of the size of the phased blocks is based on the size of the contigs generated by HiFiasm on HiC mode. We follow the quality metric nomenclature established by Rhie et al. (2021), with the genome quality code x.y.P.Q.C, where, x = log10[contig NG50]; y = log10[scaffold NG50]; P = log10 [phased block NG50]; Q = Phred base accuracy QV (quality value); and C = % genome represented by the first “n” scaffolds, following a karyotype of 2n = 36 for this species, estimated as a mode from ancestral species number of chromosomes [Genome on a Tree—GoaT; tax_name (G. wislizenii); Porter et al. 1994; Challis et al. 2023]. Quality metrics for the notation were calculated on the haplotype one assembly.
Genome annotation
We annotated the reference assembly using the NCBI Eukaryotic Genome Annotation Pipeline (hereafter, “EGAPx”) which is published in the NCBI RefSeq database (O’Leary et al. 2016) (accessible through https://github.com/ncbi/egapx). Annotation features were identified by aligning transcripts and proteins from related taxa in the RefSeq database using BLAST (Camacho et al. 2009). Novel, species-specific RNA-Seq reads generated from seven tissue types were also aligned to the assembly using the alignment software STAR (Dobin et al. 2013). Additional features are predicted using HMM-based gene models using the NCBI Gnomon software (Souvorov et al. 2010). We evaluated the quality and completeness of our annotation by the comparing the longest protein for each annotated coding gene to eukaryotes (odb12), tetrapods (odb10), and squamates (odb12) using BUSCO (Manni et al. 2021).
Results
Sequencing data
The Omni-C library generated 496.36 million read pairs and the PacBio HiFi library generated 7.75 million reads. The PacBio HiFi sequences yielded ~41X genome coverage and had an N50 read length of 13,378 bp; a minimum read length of 107 bp; a mean read length of 13,060 bp; and a maximum read length of 56,853 bp (see Supplementary Fig. S1 for read length distribution). Based on the PacBio HiFi data, Genomescope 2.0 estimated a genome size of 2.44 Gb, a 0.146% sequencing error rate, and 0.61% heterozygosity. The k-mer spectrum shows a bimodal distribution with a major coverage peak at ~ 40-fold coverage and a minor peak at ~ 20-fold coverage (Fig. 2A). Sequencing of mRNA libraries of blood, brain, intestine, liver, lung, muscle, and ovary generated 57.7, 58.1, 46.8, 67.6, 55.3, 53.3, and 55.8 M read pairs, respectively.
Visual overview of genome assembly metrics. A) K-mer spectra output generated from PacBio HiFi data without adapters using GenomeScope2.0. The bimodal pattern observed corresponds to a diploid genome. K-mers covered at lower coverage and low frequency correspond to differences between haplotypes, whereas the higher coverage and high-frequency k-mers correspond to the similarities between haplotypes. B) BlobToolkit Snail plot displaying the quality metrics presented in Table 2 for the Gambelia wislizenii primary assembly (haplotype 2; rGamWis1.0.hap2). The plot circle represents the full size of the assembly. From the inside-out, the central plot covers length-related metrics. The red line represents the size of the longest scaffold; all other scaffolds are arranged in size-order moving clockwise around the plot and drawn in gray starting from the outside of the central plot. Dark and light orange arcs show the scaffold N50 and scaffold N90 values. The central light gray spiral shows the cumulative scaffold count with a white line at each order of magnitude. White regions in this area reflect the proportion of Ns in the assembly. The dark vs. light blue area around it shows mean, maximum and minimum GS vs. AT content at 0.1% intervals (Challis et al. 2020). Omni-C contact maps for haplotype 2 C) and haplotype 1 D) genome assembly generated with PretextSnapshot. Omni-C contact maps translate proximity of genomic regions in 3D space to contiguous linear organization. Each line in the contact map corresponds to sequencing data supporting the linkage (or join) between two of such regions. Scaffolds are separated by black lines and higher density corresponds to high levels of fragmentation.
Nuclear genome assembly
The final assembly (rGamWis1) consists of two pseudo haplotypes, haplotypes 1 and 2, with both genome sizes close to the estimated value from Genomescope 2.0 (Fig. 2A: Pflug et al. 2020). Haplotype 1 assembly (rGamWis1.0.hap1) consists of 141 scaffolds spanning 2.47 Gb with contig N50 of 141.37 Mb, scaffold N50 of 379.35 Mb, longest contig of 293.11 Mb, and largest scaffold of 501.95 Mb. The haplotype 2 assembly (rGamWis1.0.hap2) consists of 69 scaffolds, spanning 2.47 Gb with contig N50 of 138.19 Mb, scaffold N50 of 380.06 Mb, largest contig 321.14 Mb, and largest scaffold of 502.04 Mb. Detailed assembly statistics are reported in Table 2, and graphical representation for the haplotype 2 assembly is shown in Fig. 2B (see Supplementary Fig. S1 for the haplotype 1 assembly).
The haplotype 1 assembly has a BUSCO completeness score of 97.4% using the Tetrapoda gene set, a per-base quality (QV) of 67.8, a k-mer completeness of 89.28% and a frameshift indel QV of 48.49; while the haplotype 2 assembly has a BUSCO completeness score of 97.4% using the same gene set, a per-base quality (QV) of 68.45, a k-mer completeness of 89.29%, and a frameshift indel QV of 48.58.
During manual curation, we generated 1 break and 11 joins on haplotype 1 and 2 breaks and 15 joins on haplotype 2. We were able to close two gaps on haplotype 2. No further contigs were modified or removed. The Omni-C contact maps show highly contiguous assemblies with chromosome-length scaffolds (Fig. 2B). The six largest scaffolds range from 163.1 to 502.1 Mb, the next 12 largest scaffolds range from 11.7 to 45.5 Mb, and all remaining scaffolds range from 10.8 to 37.5 Kb. Assembly statistics are reported in Table 2 and represented graphically in Fig. 2C. Both assemblies are now deposited on NCBI (see Table 2 and Data availability for details).
We assembled a partial mitochondrial genome from the PacBio HiFi reads using the reference-guided pipeline MitoHiFi. The final mitochondrial sequence has a size of 25,077 bp, with a base composition of A = 29.72%, C = 21.27%, G = 10.38%, and T = 29.14%, with 13 unique transfer RNAs.
Our final genome annotation included 23,279 genes, with a tetrapod BUSCO completeness of 98.9%. A list of annotation statistics and BUSCO score breakdowns for eukaryotes, tetrapods, and squamates is reported in Table 2.
Discussion
Here we present a high-quality reference genome for G. wislizenii. The excellent quality of the genome is exemplified by BUSCO completeness scores of 97.4% for both haplotypes, and on a scaffold distribution that is consistent with the known chromosome arrangement for the species. The 2n = 36 karyotype of G. wislizenii is composed of six pairs of macrochromosomes and 12 pairs of microchromosomes (Porter et al. 1994). Haplotype 2 of the G. wislizenii genome presented here includes six scaffolds that are consistent in size with macrochrosomes (163.1 to 502.1 Mb), 12 additional scaffolds that are consistent in size with microchromsomes (11.7 to 45.5 Mb), and a remaining set of 51 scaffolds that are much smaller (10.8 to 37.5 Kb) and most likely comprise difficult-to-assemble sequences rich in repeats.
There are now 217 reference genomes representing squamate reptiles uploaded onto NCBI. Of these, 40 represent the major lizard clade, Iguania, a cosmopolitan assemblage with nearly 2,100 currently recognized species (Uetz et al. 2024). Iguania is divided into 14 families, one of which is Crotaphytidae, and the G. wislizenii genome reported here is the first for this small but deeply divergent iguanian clade composed of just two genera and 12 species. More than half of the available iguanian genomes (n = 23) are concentrated among the Old World acrodontan families Agamidae (n = 13) and Chameleonidae (n = 10). Among the 12 families of pleurodont iguanians, only five of 12 families currently have a reference genome posted on NCBI: Anolidae (n = 5), Corytophanidae (n = 1), Iguanidae (n = 3), Phrynosomatidae (n = 7), and now Crotaphytidae (n = 1). The genome presented here will thus provide a valuable resource for broader comparative genomic studies spanning the hyper-diverse Iguania, as well as for studies investigating the conservation, ecology, and evolutionary history of G. wislizenii, and its close relatives within the genera Gambelia and Crotaphytus.
From a conservation perspective, we note that Gambelia includes one of the most highly endangered reptiles in the United States, G. sila, as well as one that barely enters the country at its southern border, G. copei. Having access to genomic resources across a dense sample spanning all three species will be critically important for assessing the conservation genetics, population demography, and long-term viability of these species in California. As a captive breeding and release program is already underway for G. sila, having both this reference genome and the landscape-level analysis of the species’ genomic variation will help inform breeding strategies, while maintaining the naturally occurring genomic variation that characterizes the species. These landscape genomic resources to be generated via the CCGP will also offer the potential to investigate the genetic underpinnings of important features of the biology of leopard lizards, including gravid and breeding coloration, territoriality, and reversals in patterns of sexual dimorphism, as well as the extent and nature of gene flow at Gambelia species boundaries. Understanding historical gene flow among the species will, more broadly, also shed light on landscape scale processes, such as the primordial separation of the San Joaquin and Mojave Deserts and their biotas, which in turn can inform conservation practices for many species in those biomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdennur N, Mirny LA. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36:311–316. 10.1093/bioinformatics/btz 54031290943 PMC 8205516 · doi ↗ · pubmed ↗
- 2Allio R, Schomaker-Bastos A, Romiguier J, Prosdocimi F, Nabholz B, Delsuc F. Mito Finder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020;20:892–905. 10.1111/1755-0998.1316032243090 PMC 7497042 · doi ↗ · pubmed ↗
- 3Beninde J, Toffelmier E, Shaffer HB. A brief history of population genetic research in California and an evaluation of its utility for conservation decision-making. J Hered. 2022;113:604–614. 10.1093/jhered/esac 04936056714 PMC 9709982 · doi ↗ · pubmed ↗
- 4Bishop AP, Westeen EP, Yuan ML, Escalona M, Beraut E, Fairbairn C, Marimuthu MPA, Nguyen O, Chumchim N, Toffelmier E, et al. Assembly of the largest squamate reference genome to date: the western fence lizard. J Hered. 2023;114:521–528. 10.1093/jhered/esad 03737335574 PMC 10445515 · doi ↗ · pubmed ↗
- 5Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421–421. 10.1186/1471-2105-10-42120003500 PMC 2803857 · doi ↗ · pubmed ↗
- 6Challis R, Richards E, Rajan J, Cochrane G, Blaxter M. Blob Tool Kit—interactive quality assessment of genome assemblies. G 3 (Bethesda). 2020;10:1361–1374. 10.1534/g 3.119.40090832071071 PMC 7144090 · doi ↗ · pubmed ↗
- 7Challis R, Kumar S, Sotero-Caio C, Brown M, Blaxter M. Genomes on a Tree (Goa T): A versatile, scalable search engine for genomic and sequencing project metadata across the eukaryotic tree of life. Wellcome Open Res. 2023;8:24. 10.12688/wellcomeopenres.18658.136864925 PMC 9971660 · doi ↗ · pubmed ↗
- 8Cheng H, Jarvis ED, Fedrigo O, Koepfli K-P, Urban L, Gemmell NJ, Li H. Haplotype-resolved assembly of diploid genomes without parental data. Nat Biotechnol. 2022:40:1332–1335. 10.1038/s 41587-022-01261-x PMC 946469935332338 · doi ↗ · pubmed ↗