Chinese guidelines for diagnosis and treatment of myelodysplastic neoplasms (2026)

TL;DR
This paper presents updated Chinese guidelines for diagnosing and treating myelodysplastic neoplasms, incorporating recent research and WHO classification changes.
Contribution
The paper provides revised clinical guidelines for MDS diagnosis and treatment based on recent advancements and updated WHO classifications.
Findings
MDS is a heterogeneous group of myeloid tumors with risk of progressing to acute myeloid leukemia.
Recent advances in basic research and targeted therapies have influenced the updated guidelines.
The fifth edition of the WHO classification has updated the MDS categorization.
Abstract
骨髓增生异常肿瘤(MDS)是一组起源于造血干细胞的高度异质性髓系肿瘤,临床表现为一系或多系血细胞减少,有向急性髓系白血病转化的风险。近年来,MDS在基础研究和新型靶向药物治疗方面取得了较大进展,第五版WHO分类对MDS的分型进行了更新。因此,中华医学会血液学分会对《骨髓增生异常综合征中国诊断与治疗指南(2019年版)》进行了修订,以规范MDS的诊断、鉴别诊断、预后评估及治疗选择,从而更好地指导临床实践。
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
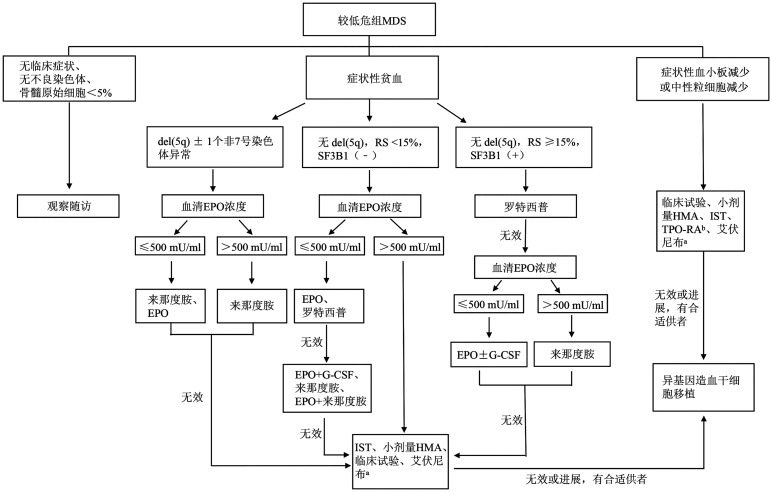 Figure 1
Figure 1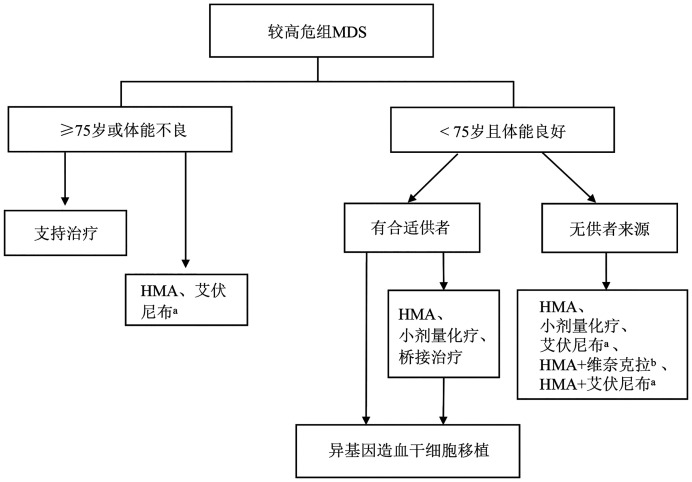 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
骨髓增生异常肿瘤(myelodysplastic neoplasms,MDS),此前称骨髓增生异常综合征(myelodysplastic syndromes,MDS),是一组起源于造血干细胞的恶性克隆性疾病,其特点是一系或多系血细胞减少、髓系各阶段细胞发育异常和高风险向急性髓系白血病(acute myeloid leukemia,AML)转化。2014–2018年间,经年龄调整的MDS每年的发病率为4.2/10万。MDS好发于老年人群,发病率随着年龄的增长显著升高,诊断时的中位年龄为77岁,小于50岁患者的比例不足10%[1]。近年来,MDS的临床研究取得了较大进展,为进一步提高我国医务人员对MDS的认识和诊治水平,中华医学会血液学分会在《骨髓增生异常综合征中国诊断与治疗指南(2019年版)》[2]的基础上制订了本版指南。
一、诊断标准与鉴别诊断
MDS的最低诊断标准见表1。按照世界卫生组织(World Health Organization,WHO)的标准,血细胞减少的标准为:中性粒细胞绝对计数(absolute neutrophil count,ANC)<1.8×10^9^/L,血红蛋白(HGB)<130 g/L(男)或<120 g/L(女),血小板计数(PLT)<150×10^9^/L。
MDS的诊断属于排除性诊断,应首先排除可能发展为MDS的前驱疾病,包括克隆性造血(clonal hematopoiesis,CH)、潜能未定克隆性造血(clonal hematopoiesis of indeterminate potential,CHIP)、意义未明特发性血细胞减少(idiopathic cytopenia of undetermined significance,ICUS)以及意义未明克隆性血细胞减少(clonal cytopenia of undetermined significance,CCUS)等[1],[4]。
CH指携带获得性突变的造血干细胞克隆性扩增,其发生率随年龄增长而增加,与全因死亡率、心血管疾病及髓系肿瘤风险相关,如VEXAS(空泡、E1酶、X连锁、自身炎症性、体细胞突变)综合征体现了炎症和CH或髓系肿瘤之间的相互作用[5]。第五版WHO分型将CH视为髓系前体细胞损伤。CHIP是CH的主要亚型,特指检测到变异等位基因频率(variant allele frequency,VAF)≥2%(男性X连锁基因≥4%)的髓系肿瘤相关基因突变,但无血细胞减少或可诊断的血液疾病。ICUS指患者在缺乏克隆性证据的情况下存在持续不明原因的血细胞减少,检测不到血液肿瘤相关驱动基因。若CHIP患者出现持续性不明原因血细胞减少,且不符合髓系肿瘤诊断标准,则称为CCUS。
其他常见需要与MDS鉴别的因素或疾病包括:
-
先天性或遗传性血液病:如先天性红细胞生成异常性贫血、遗传性铁粒幼细胞性贫血、先天性角化不良、范可尼贫血、先天性中性粒细胞减少症和先天性纯红细胞再生障碍等。
-
其他累及造血干细胞的疾病:如再生障碍性贫血、阵发性睡眠性血红蛋白尿症、原发性骨髓纤维化、大颗粒淋巴细胞白血病、急性白血病等。
-
巨幼细胞贫血:MDS患者细胞形态发育异常可见巨幼变,易与巨幼细胞贫血混淆,但后者是由叶酸、维生素B_12_缺乏所致。
-
接受细胞毒性药物、细胞因子治疗或接触有血液毒性的化学制品或生物制剂等。
-
慢性病性贫血(感染、非感染性疾病或肿瘤)、慢性肝病、慢性肾功能不全、病毒感染(如HIV、巨细胞病毒、EB病毒等)。
-
自身免疫性血细胞减少、甲状腺功能减退或其他甲状腺疾病。
-
重金属(如砷剂等)中毒、过度饮酒、铜缺乏。
二、分型
第五版WHO分型标准保留既往形态学病态造血标准,但进一步强调遗传学在MDS发病、诊断和预后中的重要作用,新提出以遗传学和形态学来分型,明确遗传学改变分型——MDS-5q、MDS-SF3B1和MDS-biTP53。新版分型去掉了基于病态造血系列的分型,统一归为MDS伴低原始细胞(MDS with low blasts,MDS-LB);在MDS-LB的基础上新增了低增生性MDS(MDS hypoplastic,MDS-h);以MDS伴原始细胞增多(MDS with increased blasts,MDS-IB)代替了前一版的MDS-EB;关注骨髓纤维化的重要性,在MDS-IB中新增了MDS-f。第五版WHO关于MDS的分型及定义见表2。
三、诊断方法
MDS的诊断基于外周血血细胞计数、骨髓和外周血涂片细胞形态学、免疫分型、细胞遗传学及分子生物学检测等,诊断时建议进行骨髓活检。
-
血象和骨髓象:约半数以上MDS患者表现为全血细胞减少。一系减少少见,多为红细胞减少。骨髓多增生活跃或明显活跃,少数病例可增生减低。病态造血的特征与MDS的生物学特性及典型遗传学改变有关,病态造血的类型归纳于表3。
-
骨髓病理检测:所有怀疑为MDS的患者均应行骨髓活检,骨髓活检细胞学分析有助于排除其他可能导致血细胞减少的因素或疾病,并提供骨髓细胞增生程度、巨核细胞数量、原始细胞比例、骨髓纤维化程度及肿瘤骨髓转移等重要信息。怀疑为MDS的患者应行Gomori银染色和原位免疫组化(immunohistochemistry,IHC),常用的检测标志包括CD34、MPO、GPA、CD61、CD42、CD68、CD20和CD3。常见病理学异常包括:红系形态及定位异常,巨核细胞胞体大小不等,核叶多变,可为单叶、双叶或多叶,可见单圆核巨核细胞及小巨核细胞;骨髓网硬蛋白纤维增生。
-
细胞遗传学检测:40%~60%的MDS患者可检出非随机的染色体异常,以−5/5q−、−7/7q−、+8、20q−和−Y最为多见。针对MDS常见异常的组套探针进行荧光原位杂交(fluorescence in situ hybridization,FISH)检测,有助于提高部分MDS患者细胞遗传学异常检出率,通常探针包括:5q31、CEP7、7q31、CEP8、20q、CEPY和TP53。MDS患者常见细胞遗传学异常见表4。
-
免疫表型:流式细胞术可用于量化CD34^+^和CD117^+^髓系祖细胞的比例,还能可靠地识别原始细胞和非原始细胞的异常特征,有助于支持MDS的诊断。欧洲白血病网/国际MDS流式细胞术工作组提出了应用流式细胞术诊断MDS的建议[6],可以根据评分不同程度地支持MDS的诊断。
-
分子遗传学检测:80%~90%的MDS患者存在基因突变。早在2017年,国内学者第一次利用二代测序揭示了我国MDS患者的基因突变谱[7],另有国内学者发现携带der(1;7)(q10;p10)的MDS患者中,RUNX1基因突变的发生频率非常高[8]。我国学者还发现,伴DDX41突变髓系肿瘤患者去甲基化治疗的缓解率达69%,两年总生存(OS)率达85%[9]。MDS国际工作组建议,基因突变监测应至少包括16个预后基因形成的17种突变类型(ASXL1、CBL、DNMT3A、ETV6、EZH2、FLT3、IDH2、KRAS、MLLPTD、NPM1、NRAS、RUNX1、SF3B15q、SF3B1α、SRSF2、TP53多重打击和U2AF1)。50岁以下患者建议加做胚系突变检测。建议MDS患者检测的体细胞突变和易感的胚系突变基因见表5。
四、预后分层
目前国内外常用的MDS疾病危险度分层的预后评分系统包括国际预后积分系统(International prognostic scoring system,IPSS)(表6)、WHO预后积分系统(WHO-based prognostic scoring system,WPSS)及修订国际预后积分系统(Revised international prognostic scoring system,IPSS-R)(表7)。国际MDS预后工作组(International Working Group for Prognosis in MDS,IWG-PM)建立了基于基因突变的预后风险模型[10],即MDS分子国际预后评分系统(Molecular International Prognosis Scoring System,IPSS-M)。该系统沿用了IPSS-R系统中的部分临床参数(包括骨髓的原始细胞比例、PLT及HGB)和细胞遗传学的危险度分组,同时筛选出31个基因(其中16个为预后相关基因形成的17种突变类型,另外15个为其他基因组)(表8)。每种突变类型被赋予不同的数据模型权重进行评分。为了简化IPSS-M的临床使用,研究开发了一个开放访问的网络版评分系统(https://mds-risk-model.com)。而国内MDS患者与国外IWG-PM队列存在以下差异:中位发病年龄更低、血细胞减少程度更严重、骨髓原始细胞增多及不良染色体核型比例增高。国内研究队列表明,IPSS-M在年龄>60岁的MDS患者中预测效力更强,提示IPSS-M的风险评估需整合年龄等相关因素以优化预测效能[11]。
五、治疗
(一)最佳支持治疗
无论患者的诊断分型、风险评分如何,对症治疗应贯穿始终。对症支持治疗的主要目标为提升患者生活质量,包括成分输血、预防及抗感染、去铁治疗。
-
成分输血:症状性贫血患者可以输注红细胞改善贫血症状。伴有出血症状的血小板减少患者输注血小板。无活动性出血的患者,推荐PLT<10×10^9^/L时予输注血小板。
-
预防及抗感染:不建议常规预防感染,但患者开始接受高强度治疗时可以预防性应用。G-CSF/GM-CSF推荐用于中性粒细胞缺乏且伴有反复或持续性感染的MDS患者,不建议持续使用。对于合并感染的患者予积极的抗感染治疗。
-
去铁治疗:慢性铁过载可对肝脏、心脏及内分泌功能尤其是胰腺功能产生不良影响。对于输血依赖的患者应定期监测血清铁蛋白(SF)水平、累计输血量和器官功能(心脏、肝脏、胰腺),评价铁过载程度(有条件的单位可采用MRI评估心脏和肝脏的铁沉积程度)。对于预期寿命≥1年、输注红细胞总量≥20 U、SF≥1 000 ng/ml至少2个月及输血依赖的患者,可实施去铁治疗,将SF降至1 000 ng/ml以下。常用的去铁药物有去铁胺和地拉罗司等。去铁胺常见的不良反应包括恶心,关节痛,肌痛,发热及注射部位疼痛、肿胀、红斑、瘙痒等,部分患者可出现肌酐升高,甚至急性肾功能衰竭。地拉罗司应用于输血依赖的MDS及中低危MDS合并铁过载,其常见的不良反应包括中性粒细胞减少和血小板减少,通常为轻度或中度。部分患者可出现肝肾功能异常,严重者可出现急性肾衰竭或肝衰竭及消化道出血。对于肌酐清除率<40 ml/min的患者,避免应用去铁胺及地拉罗司。
(二)较低危组MDS的治疗
根据现有预后评分系统,较低危组包含IPSS评分≤1分的低危组/中危-1组、IPSS-R评分≤3.5分的极低危组/低危组/中危组及IPSS-M评分≤0分的极低危组/低危组/中低危组。较低危组MDS的治疗目标是改善血象、提升患者生活质量,其治疗路径见图1。
- 促造血治疗:
(1)人促红细胞生成素(hEPO):对于存在症状性贫血或输血依赖的非del(5q)患者,hEPO应尽早使用以获得最大疗效。推荐起始剂量为每周40 000 U,连续治疗8周。若效果不佳,建议第9周起加量至每周60 000 U,继续治疗8周。对于红系应答的患者,hEPO剂量为维持应答所需要的最小剂量。若hEPO每周60 000 U红系应答仍不佳,采用EPO+G-CSF治疗8周评估疗效。hEPO治疗完成24周后,若红系应答不良,判断为hEPO治疗失败。
(2)罗特西普:是一种可溶性融合蛋白,能结合转化生长因子β(TGF-β)配体,减少SMAD2和SMAD3信号传导,从而促进幼红细胞向晚期红细胞的分化成熟。在较低危MDS中可改善红细胞的输血依赖,提升HGB水平[12]。罗特西普的安全性及有效性也在亚洲人群中得到证实[13]。罗特西普为环状铁粒幼细胞(RS)≥15%或RS≥5%伴SF3B1突变的较低危组MDS患者贫血治疗的首要选择。推荐罗特西普起始用量为每次1 mg/kg,每3周为1个疗程,后续根据治疗反应调整剂量。治疗2个疗程后若患者未脱离输血依赖,罗特西普加量至1.33 mg/kg,继续治疗2个疗程。若持续效果不佳,可调整为最大剂量1.75 mg/kg,每3周为1个疗程。罗特西普最常见的不良反应包括疲劳、腹泻、气喘、恶心和头晕,这些不良反应随着时间的推移而减少。3级以上的不良反应有骨痛及高血压,少数患者有发生静脉血栓的风险。目前我国批准罗特西普用于需要定期输注红细胞的极低危、低危和中危成人MDS的贫血治疗。
(3)TPO受体激动剂(TPO-RA):包括艾曲泊帕、海曲泊帕、阿伐曲泊帕、芦曲泊帕及罗普司亭。TPO-RA可以减少较低危MDS患者的出血风险。对于严重单一血小板减少的患者,若骨髓原始细胞<5%,可考虑单独使用TPO-RA。但需要关注TPO-RA导致的MDS疾病进展风险,需谨慎用药。目前TPO-RA尚未在MDS血小板减少患者中获批适应证。
(4)其他治疗方案:全反式维甲酸(ATRA)联合hEPO和十一酸睾酮治疗低危MDS患者,可改善红系应答及输血依赖[14]。罗特西普单药治疗复发难治低危MDS,可改善红系应答反应[15]。
-
免疫调节治疗:常用的免疫调节药物为来那度胺。对于del(5q)亚型的症状性贫血,治疗首选来那度胺。来那度胺可使部分患者减轻或脱离输血依赖,并获得细胞遗传学缓解,生存期延长。来那度胺用量为:10 mg/d,连用21 d,每28天为1个疗程,持续治疗2~4个月以评估疗效。来那度胺治疗3~6个月后HGB升高<15 g/L或红细胞输注依赖无改善,提示治疗无效。来那度胺最常见的≥3级不良反应为中性粒细胞减少和血小板减少。因此,10 mg/d×21 d方案适用于PLT>50×10^9^/L及ANC>0.5×10^9^/L的患者。对于伴有del(5q)但无输血依赖的较低危MDS患者,5 mg/d的小剂量来那度胺可降低患者发生输血依赖的风险,80%患者达到细胞遗传学反应,70%患者获得细胞遗传学的完全缓解(CR)。伴有del(5q)的MDS患者如出现下列情况不建议应用来那度胺:①骨髓原始细胞比例>5%;②合并复杂染色体核型或染色体−7;③较高危组MDS;④合并TP53基因突变。对于上述情况,推荐采用较高危组MDS的治疗方案。来那度胺在其他亚型中的应用详见下文。
-
免疫抑制治疗(IST):IST包括抗胸腺细胞球蛋白(ATG)±环孢素A,可考虑用于具备下列特征的较低危患者:年龄≤60岁、骨髓原始细胞比例≤5%、低增生MDS、正常核型或单纯染色体+8、存在输血依赖、HLA-DR15阳性、阵发性睡眠性血红蛋白尿症克隆阳性及合并STAT3突变。环孢素A起始剂量为3~5 mg·kg^−1^·d^−1^,空腹血药浓度维持在100 µmol/L以上,持续6个月。ATG剂量参考再生障碍性贫血用法。对于合并血小板减少患者,可采用IST±TPO-RA。
-
症状性贫血伴del(5q)亚型:若血清促红细胞生成素(sEPO)浓度≤500 mU/ml,首先推荐来那度胺,其次推荐hEPO。若上述治疗无效,治疗参考下文中sEPO浓度>500 mU/ml但来那度胺疗效不佳患者的治疗路径。
若sEPO浓度>500 mU/ml,首先推荐来那度胺治疗。若疗效不佳,患者合并IST使用特征时,可采用IST±TPO-RA。以上治疗效果欠佳的患者建议采用小剂量去甲基化治疗、参加临床试验或接受异基因造血干细胞移植(allo-HSCT),也可根据异柠檬酸脱氢酶1(IDH1)基因突变状态决定是否联合艾伏尼布治疗。
-
症状性贫血伴SF3B1突变亚型:首选罗特西普治疗。若罗特西普疗效不佳,根据sEPO浓度决定治疗方案。若sEPO浓度≤500 mU/ml,推荐hEPO±G-CSF。若sEPO浓度>500 mU/ml,建议接受来那度胺治疗。若以上治疗均无明显应答,考虑IST或依据IDH1基因突变状态决定是否联合艾伏尼布靶向治疗。若以上治疗应答仍不佳,建议参加临床试验或接受allo-HSCT。
-
症状性贫血伴非del(5q)、非SF3B1突变亚型:若sEPO浓度≤500 mU/ml,首先推荐采用hEPO治疗,也可采用罗特西普治疗。其他推荐包含hEPO+G-CSF、来那度胺、hEPO+来那度胺。若sEPO浓度>500 mU/ml,可根据疾病特征接受IST±TPO-RA。也可尝试hEPO联合ATRA及十一酸睾酮三药组合。建议参加临床试验、接受去甲基化治疗、接受罗特西普治疗。若效果仍不佳,可根据IDH1基因突变状态决定是否联合艾伏尼布靶向治疗或allo-HSCT。
-
症状性血小板减少或中性粒细胞减少:优先考虑临床试验。也可尝试小剂量地西他滨(Decitabine,DAC)或阿扎胞苷(Azacitidine,AZA)治疗,具体为:DAC 20 mg·m^−2^·d^−1^×3 d或AZA 75 mg·m^−2^·d^−1^×3 d,每4周为1个疗程。适合接受IST的患者,也可考虑接受IST±TPO-RA治疗。严重的单一血小板减少患者可考虑单独应用TPO-RA,但需警惕MDS白血病转化风险。对于存在IDH1基因突变的患者,可联合艾伏尼布分子靶向治疗。若以上治疗无效或发生疾病进展,考虑其他临床试验或allo-HSCT。
(三)较高危组MDS的治疗
较高危组包含IPSS评分>1分的中危-1组/高危组、IPSS-R评分>3.5分的中危组/高危组/极高危组及IPSS-M评分>0分的中高危组/高危组/极高危组。较高危组MDS的治疗目标是延缓疾病进展、延长患者生存期甚至治愈,其治疗路径见图2。
- 去甲基化治疗:推荐用于不适合造血干细胞移植的较高危MDS患者。与支持治疗组相比,去甲基化药物治疗组可降低患者向AML进展的风险、改善患者的生存。常用药物包括AZA及DAC。AZA及DAC的有效性及安全性也在国内多中心的注册研究中得到证实[16]–[17]。
(1)AZA:推荐用法为75 mg·m^−2^·d^−1^×7 d,皮下注射,每4周为1个疗程。接受AZA治疗的MDS患者,首次获得治疗反应的中位时间为3个疗程,约90%治疗有效的患者在6个疗程内获得治疗反应。因此,推荐MDS患者接受至少6个疗程AZA后评价治疗反应,有效患者可持续使用。
(2)DAC:推荐用法为20 mg·m^−2^·d^−1^×5 d,每4周为1个疗程,至少接受4个疗程后评价治疗反应,有效患者可持续使用。
(3)其他联合治疗方案:已有研究报道,接受ATRA联合DAC治疗的较高危MDS患者的总有效率、CR率及无进展生存率均显著高于接受DAC单药治疗患者[18]。
-
AML样治疗及小剂量化疗:AML样治疗适用于年龄<70岁、体能良好、骨髓原始细胞比例≥10%患者的移植前桥接治疗,也适用于不适合移植的较高危患者或复发难治患者。国内常用的治疗方案为VA方案,具体为:AZA 75 mg/m^2^,第1~7天;维奈克拉100 mg第1天,200 mg第2天,自第3天开始400 mg,应用时间依据血象调整,一般应用7~14 d。最新的Ⅲ期临床试验对比AZA联合维奈克拉与AZA单药治疗高危MDS的疗效,患者的OS率无明显差异。对于复发难治患者,鼓励参加临床试验。小剂量化疗方案为:阿糖胞苷20 mg·m^−2^·d^−1^,连用10~14 d,每4周为1个疗程。对于存在预后不良染色体核型的患者,小剂量化疗的疗效劣于AZA去甲基化治疗。
-
分子靶向治疗:
(1)BCL-2抑制剂:新型靶向药物B细胞淋巴瘤2(BCL-2)抑制剂维奈克拉与去甲基化治疗或靶向IDH1抑制剂联合用于高危难治MDS患者,在前期的临床试验中表现出良好的效果。在一项应用AZA联合维奈克拉治疗高危MDS的Ⅰb期临床试验中,维奈克拉的用量为400 mg每日1次×14 d;AZA 75 mg·m^−2^·d^−1^×7 d,每4周为1个疗程。纳入分析的107例患者中,29.9%达CR,中位OS期为26个月[19]。对于高危MDS,一项Ⅲ期临床试验表明,患者接受AZA联合维奈克拉治疗的OS率较接受AZA单药治疗无显著提高。鼓励患者参加包含维奈克拉的临床试验,但需密切关注药物相关的骨髓抑制。维奈克拉最常见的≥3级不良反应是中性粒细胞减少和血小板减少。
(2)IDH1突变抑制剂:约4%的MDS患者存在IDH1基因突变。IDH1突变抑制剂艾伏尼布在国内已获批治疗伴IDH1突变的复发难治AML[20]。一项Ⅰ期试验评估了艾伏尼布对伴IDH1基因突变的复发难治MDS患者疗效和安全性。共入组19例患者,纳入疗效评估的18例患者中,艾伏尼布的CR率+部分缓解率为38.9%。达CR患者中,68.6%患者缓解持续时间超过5年,中位OS期35.7个月。71.4%需要输注红细胞及75.0%需要输注血小板的患者脱离了输血依赖。2024年FDA批准艾伏尼布治疗伴IDH1突变的复发难治MDS。艾伏尼布常见的≥3级不良反应为疲劳和低钠血症。目前主要用于临床试验,可与去甲基化治疗联合应用,常见的不良反应主要为骨髓抑制,包括中性粒细胞减少、血小板减少、贫血等。
- allo-HSCT:allo-HSCT是目前唯一能根治MDS的治疗手段,造血干细胞来源包括同胞全相合供者、HLA匹配的无关供者、单倍型相合的供者或脐带血。allo-HSCT的适应证为:①年龄<75岁、造血干细胞移植合并症指数(HCT-CI)评分0~2分、Karnofsky体能状态(KPS)评分≥80%,MDS特异性衰弱指数(FI)评分<0.3的较高危组MDS患者;②年龄<75岁、伴严重血细胞减少、其他治疗无效的较低危组患者。
近年研究发现,胚系突变如CEBPA、DDX41、TP53、RUNX1、ANKRD26、ETV6、GATA2、SRP72、SAMD9、SAMD9L、BLM等可导致发生MDS及AML的风险升高。对于拟行移植的MDS患者,若存在以下特征之一:确诊年龄<50岁、合并≥1种其他肿瘤、体细胞突变合并潜在致病性胚系突变、疑似家族性肿瘤综合征患者,应对供者及受者的胚系突变进行检测,从而优化供者选择。移植前若骨髓原始细胞比例≥10%,是否行桥接治疗(去甲基化治疗、化疗或分子靶向治疗)仍有争议。对于年龄≤55岁的移植患者,推荐清髓性预处理方案。对于年龄>55岁,一般状态或脏器功能差的患者可使用减低剂量或减低毒性的预处理方案。对于较高危患者,尤其是IPSS-M或IPSS-R评分高危或极高危患者,尽早移植能给患者带来生存获益。MDS合并TP53突变,尤其是双等位基因失活患者,即使接受移植,总体预后也很差。若桥接治疗后骨髓原始细胞比例仍>5%,不应为降低肿瘤负荷而耽误移植的进行。
六、疗效评估
MDS国际工作组(International Working Group,IWG)于2000年提出国际统一疗效标准,2006年进行了修订,2018年和2023年分别对血液学改善和高危MDS的疗效评估标准进行更新[21]–[24]。2023版IWG标准考虑了最新的MDS分子评分系统IPSS-M,强调了细胞遗传学缓解在疗效评估中的重要性,首次提出等同于CR(CR equivalent)标准,剔除了原标准中的骨髓CR(mCR)标准,并对MDS治疗后血象恢复程度有了更精细化的分层评估。新标准同时剔除了原标准中的疾病稳定(SD)及无病生存(DFS)。2006版及2023版IWG评估标准见表9[24]。
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1WHO Classification of Tumours Editorial Board Haematolymphoid Tumours[M]5th ed, Volume 11Lyon, France IARC Press 2024
- 2中华医学会血液学分会骨髓增生异常综合征中国诊断与治疗指南(2019年版)[J]中华血液学杂志2019402899710.3760/cma.j.issn.0253-2727.2019.02.001 · doi ↗
- 3Valent P Orazi A Steensma DP Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions[J]Oncotarget 2017843734837350010.18632/oncotarget.1900829088721 PMC 5650276 · doi ↗ · pubmed ↗
- 4National Comprehensive Cancer Network NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) Myelodysplastic Syndromes (Version 1. 2026)[EB/OL](2025-10-19) [2025-12-07]. https://www.nccn.org/professionals/physician_gls/pdf/mds.pdf
- 5Beck DB Ferrada MA Sikora KA Somatic mutations in UBA 1 and severe adult-onset autoinflammatory disease[J]N Engl J Med 2020383272628263810.1056/NEJ Moa 202683433108101 PMC 7847551 · doi ↗ · pubmed ↗
- 6Porwit A BénéMC Duetz C Multiparameter flow cytometry in the evaluation of myelodysplasia: Analytical issues: Recommendations from the European Leukemia Net/International Myelodysplastic Syndrome Flow Cytometry Working Group[J]Cytometry B Clin Cytom 20231041275010.1002/cyto.b.2210836537621 PMC 10107708 · doi ↗ · pubmed ↗
- 7李冰 王静雅 刘晋琴 靶向测序检测511例骨髓增生异常综合征患者基因突变[J]中华血液学杂志201738121012101610.3760/cma.j.issn.0253-2727.2017.12.00229365392 PMC 7342197 · doi ↗ · pubmed ↗
- 8Zhang T Xu Y Pan J High frequency of RUNX 1 mutation in myelodysplastic syndrome patients with whole-arm translocation of der(1;7)(q 10;p 10)[J]Leukemia 201731102257226010.1038/leu.2017.22828720763 · doi ↗ · pubmed ↗