Export of miRNAs from activated macrophages is cooperative and HuR‐dependent
Syamantak Ghosh, Kamalika Mukherjee, Suvendra N. Bhattacharyya

TL;DR
The study reveals that activated macrophages export miRNAs in a cooperative way, facilitated by the protein HuR, which helps regulate gene repression and inflammation.
Contribution
The study identifies HuR as a key driver of cooperative miRNA export in macrophages, enabling coordinated release of miRNAs.
Findings
Activated macrophages export miRNAs cooperatively, with miR-122 enhancing the export of miR-146a and others.
HuR binds to miRNAs and promotes their entry into endosomes, facilitating cooperative export.
Cooperative miRNA export leads to synchronized expression of pro-inflammatory genes in macrophages.
Abstract
MiRNA export is a tightly regulated process crucial for maintaining balanced miRNA and target gene expression levels in metazoan cells. RNA‐interacting proteins such as HuR play a key role in the selectivity and specificity of miRNA export, thereby enabling context‐dependent release of gene‐repressing miRNAs from mammalian cells. Our results demonstrate that activated macrophages cooperatively export miRNAs, where hepatic miR‐122 significantly enhances the export of miR‐146a and other miRNAs. We also observe that this cooperative export causes a synchronized increase in the expression of pro‐inflammatory target genes in activated macrophages. In investigating the molecular mechanisms, we found that the miRNA‐binding protein HuR cooperatively binds to miRNAs and promotes their entry into endosomes, thereby facilitating their cooperative export. This highlights the selective and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
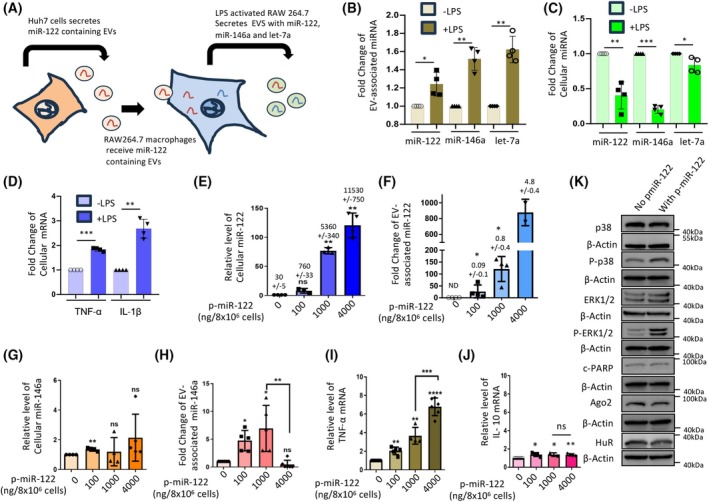 Fig. 1
Fig. 1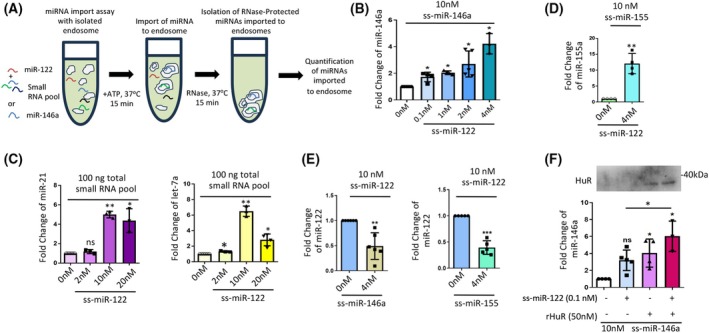 Fig. 2
Fig. 2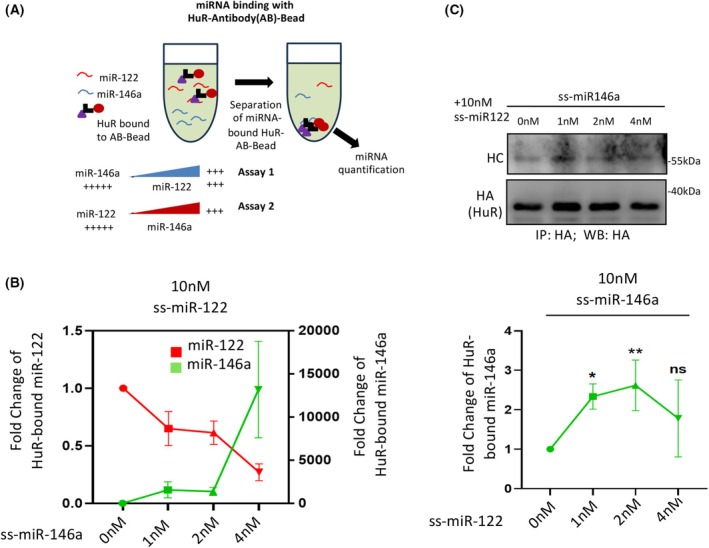 Fig. 3
Fig. 3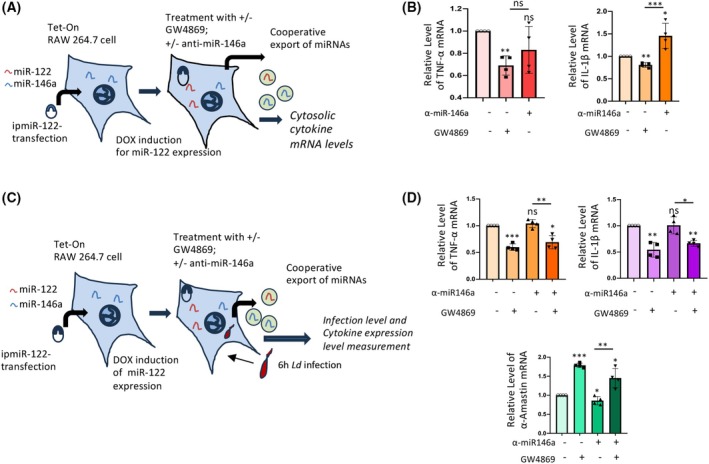 Fig. 4
Fig. 4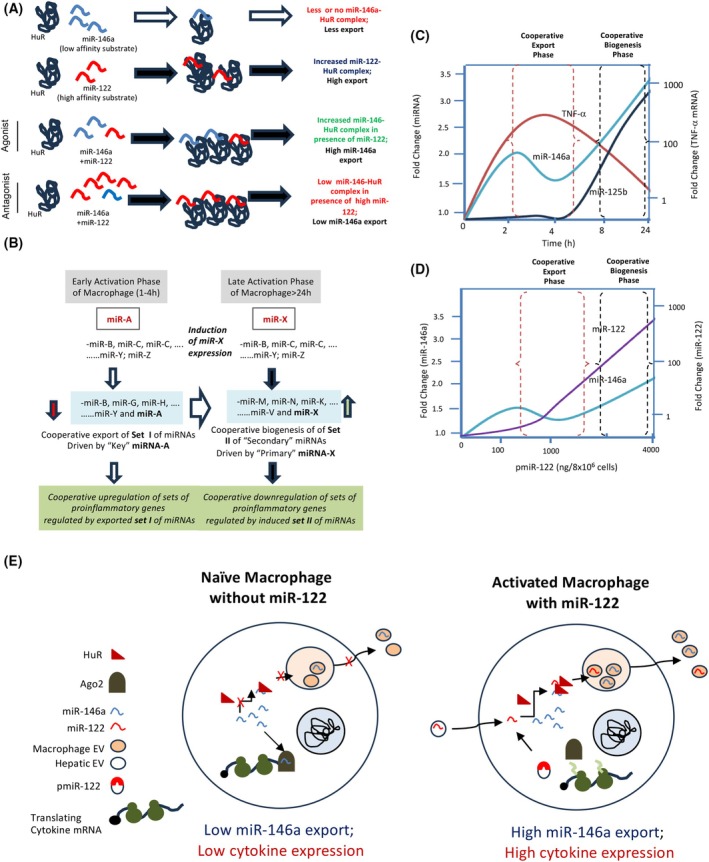 Fig. 5
Fig. 5- —College of Medicine, University of Nebraska Medical Center10.13039/100008291
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicroRNA in disease regulation · RNA Research and Splicing · Nuclear Structure and Function
Abbreviations
Ago2, Argonaute 2
ALIX, ALG‐2‐interacting protein X
ATCC, the American Type Culture Collection
ATP, adenosine tri‐phosphate
ER, endoplasmic reticulum
ESCRT, endosomal sorting complexes required for transport
EV, extracellular vesicles
FH, FLAG‐HA; FLAG (DYKDDDDK) and an HA (YPYDVPDYA) epitope
GW182, GW‐enriched protein 182
HA, hemagglutinin epitope
HRS, hepatocyte growth factor‐regulated tyrosine kinase substrate
HuR, human antigen R
miRNA, microRNA
NTA, nanoparticle tracking analysis
qRT‐PCR, quantitative real‐time reverse transcription polymerase chain reaction
RalA, Ras‐related protein A
siRNA, small interference RNA
STX5, Syntaxin 5
MicroRNAs (miRNAs), 22‐nucleotide noncoding RNAs, play a pivotal role in the precise regulation of gene expression, significantly influencing temporal and spatial changes happening at the post‐transcriptional level [1, 2, 3]. By forming imperfect base‐pairing interactions with target mRNA sequences, miRNAs induce translational repression and facilitate the degradation of these repressed messages [4, 5].
Extracellular export is critical for regulating the functional miRNA pool and for communicating miRNA between neighboring cells, thereby balancing miRNA activity in both donor and recipient cells [6, 7, 8]. In addition to Ago‐ or RNA‐binding protein nucleophosmin‐1 [9] or HDL‐associated [10] miRNA export, vesicle‐mediated export of specific miRNAs from a donor cell helps lower intracellular miRNA levels, enabling cells to refine their gene expression profiles and adapt to changing conditions, such as metabolic stress [11]. Recent studies have shown that extracellular vesicles (EVs) transport RNA and protein cargo across cellular membranes [12, 13]. The selective export of miRNAs via EVs is influenced by intrinsic and extrinsic factors that, in turn, modulate miRNA activity in higher eukaryotes, with significant implications for cellular physiology and disease initiation [14]. Interactions with specific RNA‐binding proteins, including the La antigen, YBX1, or HuR, dictate the export of miRNAs via EVs to neighboring cells [6, 7, 11, 15, 16, 17, 18, 19]. These proteins selectively enhance miRNA export, thereby regulating cytosolic miRNA levels. In addition to HuR and YBX1, many other RNA‐binding proteins have been identified that regulate miRNA export across various mammalian cell types.
The endosomal membrane is a critical location for the sorting and packaging of miRNAs into nascent exosomes or intraluminal vesicles (ILVs) [20]. An innovative in vitro assay system has recently been developed by our group to study miRNA entry into endosomes [21]. This system has proven invaluable for understanding the selective entry of miRNAs into endosomes, a prerequisite for their export via EVs in mammalian cells. RNA‐binding proteins play a key role in miRNA export and endosomal targeting. Two RNA‐binding proteins, Alyref and Fus, are involved in the export of miRNAs carrying the strongest EXOmotif, CGGGAG [6]. The role of hnRNPA2B1 in miRNA entry into endosomes has been investigated previously [17]. Modification of miRNA, like methylation, causes selective entry of m6A‐modified miRNAs into EVs for export in an hnRNPA2B1‐dependent manner [22]. A recent study also highlights the RNA‐binding protein PCBP2, which impairs SYNCRIP‐dependent miRNA loading by interacting with cellular retention motifs of miRNAs and functions antagonistically in miRNA regulation [23]. One pertinent question regarding miRNA export regulation concerns whether antagonistic or cooperative interactions occur between miRNAs during extracellular export.
Observations indicate that miR‐122 is exported from hepatic cells upon exposure to elevated lipid levels, thereby inducing inflammation in resident tissue macrophages, particularly Kupffer cells, in the murine liver [24, 25]. Notably, liver macrophages initiate the expression of inflammatory cytokines upon receiving EVs from hepatic cells that carry miR‐122 as cargo [25]. Furthermore, LPS‐mediated macrophage activation also leads to the secretion of specific miRNAs required for macrophage activation. It is also noteworthy that restricted miRNA export is linked to impaired macrophage activation in LPS‐treated cells [26].
Cytokine expression is regulated at the pre‐ and post‐transcriptional levels in activated macrophages, and miRNAs play a key role in modulating cytokine mRNA stability and translatability [27, 28]. miR‐146a is induced in macrophages during LPS activation [29] and regulated export of miR‐146a ensures regulation of inflammatory responses in pathogen ‐invaded macrophages [30].
In this manuscript, we documented EV‐mediated upregulation of export of miR‐146a in macrophages expressing miR‐122. The export of miR‐146a was found to be essential for miR‐122‐induced macrophage activation. Observations suggest that miR‐146a is cooperatively packaged into endosomes, a process influenced by miR‐122 in both ex vivo and in vitro contexts. We also observe that HuR, a strong and reversible binder of miR‐122 [11, 31], can bind to a low‐affinity substrate, miR‐146a, in the presence of miR‐122. This observation supports the notion of a cooperative binding mechanism between HuR and miRNAs, suggesting that by interacting with miRNA substrates, HuR ensures cooperative miRNA export from activated macrophages, driven by HuR‐mediated entry of miRNAs into endosomes.
In summary, during macrophage activation, the cooperative interaction between the stress‐response protein HuR and miRNAs promotes increased export of specific miRNAs. This process is initiated by the presence of one or more miRNAs with a strong affinity for HuR to increase the export of secondary miRNAs that are otherwise poor binders of HuR due to their low affinity. These cooperative interactions facilitate endosomal entry and simultaneous export of these secondary miRNAs, which are crucial for activating and inflammatory cytokines expression in mammalian macrophage cells.
Materials and methods
Cell culture and cell transfections
HEK293 (CRL‐1573; RRID CVCL_0045), RAW264.7 (TIB‐71; RRID CVCL_0493), and C6 glioblastoma (CCL‐107; RRID CVCL_1074) cells were obtained from ATTC and cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Waltham, MA, USA) supplemented with 10% heat‐inactivated fetal calf serum (HI‐FCS, Gibco) and 1% Penstrep (Gibco). RAW264.7 murine macrophage cells were grown in RPMI1640 medium (Gibco) containing 2 mm L‐glutamine and 0.5% β‐mercaptoethanol, along with 10% FBS and 1% Penstrep. All cells were maintained at 37 °C in a 5% CO_2_ incubator. RAW264.7 cells were stimulated with LPS (10 ng·mL^−1^ for 4 h) derived from Escherichia coli O111:B4 (Calbiochem) to activate macrophages. For EV‐free growth medium, 10% EV‐depleted FBS, prepared by ultracentrifugation at 100 000 ** g ** for 4 h, was added to DMEM or RPMI‐1640 as appropriate. Transfections of plasmids and cotransfections with miRNA inhibitors (anti‐miR oligonucleotides, 30 nm) in HEK293 and C6 cells were performed using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) following the manufacturer's protocol. Details of plasmids and chemicals are listed in Table S1.
Cell line authentication and mycoplasma testing
All cell lines used in this study have been authenticated via short tandem repeat (STR) profiling within the past three years (since February 2023). Additionally, each cell line was tested for mycoplasma contamination using the MycoAlert® PLUS Mycoplasma Detection Kit (Lonza, Switzerland) and confirmed negative prior to the experiments.
Extracellular vesicles isolation and characterization
Huh7, and RAW264.7 cells transfected with specific plasmids were split 24‐h post‐transfection into 90 mm plates and incubated for 24 h to achieve 70–80% confluency. The conditioned media were subjected to EV isolation as described in a previous report, with minor modifications [32]. Cells were cultured in growth medium with serum depleted of EVs to avoid interference from exosomes or EVs in the serum. The conditioned media were cleared of cellular debris and contaminants by centrifugation at 2000 ** g ** for 10 min and 10 000 ** g ** for 30 min, then filtered through a 0.22‐μm filter for additional purification. EVs were isolated by ultracentrifugation of the clarified media at 100 000 ** g ** for 90 min. When protein analysis was required, an additional ultracentrifugation step was performed on a 30% sucrose cushion at 100 000 ** g ** for 90 min. Afterward, the EV layer was diluted with 1× PBS and ultracentrifuged again at 100 000 ** g ** for 90 min to obtain a pellet. For characterization, EVs were diluted 10‐fold with PBS, and 1 mL of this suspension was analyzed using a Nanoparticle Tracker (Nanosight NS‐300) to measure EV number, size, and other parameters [21].
EV treatment of recipient cells
After EV isolation, the pellet was resuspended in fresh RPMI‐1640 with 10% EV–depleted FBS and 1% Pen‐Strep, then filtered through a 0.22‐μm filter. The EVs were added to the recipient macrophage cell line for 24 h. Subsequently, macrophages received a 10 ng·mL^−1^ LPS treatment for 4 h, 20 h after EV exposure. For treatment, EVs derived from 15 × 10^6^ cells (from five 60 mm plates) were added to 2.4 × 10^6^ recipient cells (from a single 60 mm plate) [30].
Parasite culture and infection to macrophage cell line
The Leishmania donovani (Ld) strain AG83 (MAOM/IN/83/AG83), derived from an Indian Kala‐azar patient, was maintained in golden hamsters [33]. All animal experiments were performed in accordance with the National Regulatory Guidelines issued by the Committee for Supervision of Experiments on Animals, Ministry of Environment and Forest, Government of India. These experiments were performed following the protocol approved by the Institutional Animal Ethics Committee by CSIR‐Indian Institute of Chemical Biology, India. Amastigotes were isolated from infected hamster spleens and converted into promastigotes. These promastigotes were cultured in M199 medium (Gibco) with 10% FBS (Gibco) and 1% Pen‐Strep (Gibco) at 22 °C. RAW264.7 cells were then infected with stationary‐phase Ld promastigotes from the second to fourth passage at a 1 : 10 ratio (cells to Ld) for 6 h [30].
Optiprep density gradient centrifugation
For subcellular organelle fractionation, a 3–30% continuous gradient was created using Optiprep density gradient medium with 60% iodixanol solution (Sigma‐Aldrich, St. Louis, MO, USA) in a buffer containing 78 mm KCl, 4 mm MgCl_2_, 8.4 mm CaCl_2_, 10 mm EGTA, and 50 mm HEPES (pH 7.0). Cells were washed with cold 1× PBS before lysis with a Dounce homogenizer (Kontes glass) in a buffer of 0.25 m sucrose, 78 mm KCl, 4 mm MgCl_2_, 8.4 mm CaCl_2_, 10 mm EGTA, and 50 mm HEPES (pH 7.0), supplemented with 100 μg·mL^−1^ cycloheximide, 0.5 mm DTT, and 1× PMSF. Two centrifugations were performed at 1000 ** g ** for 5 min, after which the cell lysate was gently layered onto the gradient. Ultracentrifugation was performed for 5 h at 133 000 ** g ** using a SW60‐Ti rotor (Beckman Coulter, Switzerland) to separate the layers. After centrifugation, ten fractions were collected from the top for subsequent protein and RNA analysis [21].
Endosome enrichment by subcellular fractionation
Optiprep density gradient medium, consisting of 60% iodixanol solution (Sigma‐Aldrich), was used to prepare a 3 mL step gradient with 5–10–15% layers. This involved gently overlaying 1 mL of lighter solution over 1 mL of heavier gradient in a buffer containing 78 mm KCl, 4 mm MgCl_2_, 8.4 mm CaCl_2_, 10 mm EGTA, and 50 mm HEPES (pH 7.0) for subcellular organelle separation. Cells were washed with 1× PBS and homogenized using a Dounce homogenizer (Kontes glass) in a buffer with 0.25 m sucrose, 78 mm KCl, 4 mm MgCl_2_, 8.4 mm CaCl_2_, 10 mm EGTA, and 50 mm HEPES (pH 7.0), supplemented with 100 μg·mL^−1^ cycloheximide, 0.5 mm DTT, and 1× PMSF. The lysate was clarified by two centrifugations at 1000 ** g ** for 5 min each, then 1 mL of the clarified lysate was layered atop the freshly prepared 3 mL gradient at 4 °C. The tubes were ultracentrifuged at 133 000 ** g ** for 5 h to separate the gradient, and seven equal‐volume fractions were collected from the top for endosome/MVB isolation and subsequent protein and RNA analysis. For endosome enrichment, the top three fractions were pooled and diluted with a buffer containing 78 mm KCl, 4 mm MgCl_2_, 8.4 mm CaCl_2_, 10 mm EGTA, and 50 mm HEPES (pH 7.0) to a final volume of 4 mL, then ultracentrifuged again at 133 000 ** g ** for 2 h. After centrifugation, the membrane pellet enriched with endosomes was resuspended in a buffer with 0.25 m sucrose, 78 mm KCl, 4 mm MgCl_2_, 8.4 mm CaCl_2_, 10 mm EGTA, and 50 mm HEPES (pH 7.0), supplemented with 100 μg·mL^−1^ cycloheximide, 0.5 mm DTT, and 1× PMSF. The resulting endosomal suspension was kept on ice before performing the in vitro assay [21].
Cell‐free in vitro reconstitution assay
The cell‐free in vitro assays using endosomes were performed according to a previously published protocol [34]. Endosome‐enriched suspensions from either HEK293 or C6 cells were equally divided for different experimental groups. In line with the experiments detailed in the Results section, synthetic miRNA and small RNA pools were used as cargo in the presence of ATP. Incubation was conducted for 30 min at 37 °C, after which an RNase protection assay was performed as described in a previous publication [21]. Vesicles were washed, re‐isolated by ultracentrifugation at 133 000 ** g ** for 2 h, then processed for RNA and protein analysis.
RNA isolation and real‐time PCR
Total RNA was extracted using TRIzol or TRIzol LS reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions. Samples with low miRNA content were precipitated with isopropanol and the glycoblue coprecipitant (Thermo Fisher Scientific, Waltham, MA, USA). cDNA was synthesized from 100 to 200 ng of RNA from cellular samples, or from equal volumes of RNA from in vitro or EV samples, using a TaqMan reverse transcription kit (Applied Biosystems, Carlsbad, CA, USA). miRNA levels were measured by real‐time PCR using specific primers. For quantification, two‐step RT‐PCR was performed on a Bio‐Rad CFX96TM system with TaqMan‐based miRNA assays. One‐third of the reverse transcription mix was used for PCR amplification with TaqMan Universal PCR Master Mix No AmpErase (Applied Biosystems) and corresponding TaqMan reagents. Analyses were done in triplicate. U6 snRNA‐normalized cellular miRNA levels were measured. For in vitro samples with synthetic miR‐122, endosomal miR‐146a was used to normalize miR‐122. For mRNA quantification, 200 ng of total RNA was reverse‐transcribed and analyzed by qPCR using SYBR Green (Eurogentec, Belgium). mRNA levels were normalized to GAPDH, with each sample run in triplicate using the comparative Ct method [21]. Details of the miRNA assay ID are listed in Table S2. The list and sequence of oligonucleotides and anti‐miRs are listed in Tables S3 and S4.
Immunoblotting
Protein western blotting was performed as described in previous reports [34, 35]. Protein samples, including cellular lysates, subcellular fractions, or immunoprecipitated proteins, were separated by SDS‐polyacrylamide gel electrophoresis and then transferred to PVDF nylon membranes overnight at 4 °C. The membranes were blocked in 3% BSA for 1 h, then probed with specific antibodies for at least 16 h at 4 °C. Afterward, the membranes were incubated for 1 h at room temperature with horseradish peroxidase‐conjugated secondary antibodies (diluted 1:8000). Images of the developed western blots were captured using a UVP BioImager 600 system with VisionWorks Life Science software, version 6.8 [11]. Details of antibodies are listed in Table S5.
Cooperative miRNA association/dissociation kinetics assay
For the cooperative miRNA association and dissociation kinetics assay, HEK293 cells were lysed using a buffer (20 mm Tris/HCl pH 7.5, 150 mm KCl, 5 mm MgCl_2_, 1 mm DTT) containing 0.5% Triton X‐100, 0.5% sodium deoxycholate, 40 U·mL^−1^ RNase inhibitor, and 1× PMSF for 30 min at 4 °C. The cells were then sonicated with three pulses of 10 s each. Lysates were clarified by centrifugation at 16 000 ** g ** for 15 min. Protein‐G agarose beads were blocked with 5% BSA in 1× lysis buffer for 1 h, then washed three times with 1× IP/wash buffer (20 mm Tris/HCl pH 7.5, 150 mm KCl, 5 mm MgCl_2_, 1 mm DTT). The beads were incubated with a specific antibody in 1× lysis buffer (final dilution 1:100) for 3 h at 4 °C. The clarified lysates were incubated with an anti‐HuR antibody bound to Protein‐G agarose beads, rotated overnight at 4 °C. The beads were washed three times and resuspended in an assay buffer (20 mm Tris pH 7.5, 5 mm MgCl_2_, 150 mm KCl, 1× PMSF, 2 mm DTT, 40 U·mL^−1^ RNase inhibitor). Synthetic miRNAs were added to the reaction mix as described in the results. The reaction was performed at room temperature for 15 min with shaking. Afterward, the beads were washed three times, divided into two equal parts, and each was analyzed for bound proteins and RNAs by western blots and qRT‐PCR, respectively.
Small RNA pool isolation
Small RNA was extracted from cellular lysate with the miRVANA miRNA isolation kit (Thermo Fisher Scientific), following the manufacturer's instructions. This small RNA pool served as the cargo in in vitro assay reactions, and the packaging levels of individual miRNAs were subsequently analyzed [21].
Phosphate 5′ end‐labeling of synthetic miRNA
Essentially, this was done by following a published protocol [21]. Fifty picomoles of synthetic miR‐122 RNA (22 nt) were incubated with T4 Polynucleotide Kinase (PNK) (10 units·μL^−1^) and 1 mm ATP in 1× T4 PNK buffer at 37 °C for 30 min under static conditions. Reactions were stopped with Tris‐EDTA, filtered through a mini Quick spin oligo column (Roche Diagnostics, Germany). RNA was extracted with TriZol LS and CHCl3, precipitated with isopropanol at −20 °C overnight with glycoblue coprecipitant. The pellet was resuspended in nuclease‐free water to a final concentration of 1 pmole·μL^−1^.
Preparation of recombinant HuR
Recombinant HuR protein was purified following the detailed procedure previously outlined [11]. For HuR expression in E. coli, the construct containing the HuR coding region in pET42a(+) was used. The protein was produced in BL21DE3 E. coli cells that are optimized for codon Usage. IPTG‐induced overnight cultures of E. coli BL21 were lysed by incubation with a lysis buffer composed of 20 mm Tris/HCl (pH 7.5), 300 mm KCl, 2 mm MgCl_2_, 5 mm β‐mercaptoethanol, 50 mm imidazole, 0.5% Triton X‐100, 5% glycerol, 0.5 mg·mL^−1^ lysozyme, and a protease inhibitor cocktail (Roche, Germany) without EDTA for 30 min, followed by sonication for three 10‐s pulses. The resulting lysate was clarified and incubated with pre‐equilibrated Ni‐NTA agarose beads (Qiagen, Germantown, MD, USA) for 4 h at 4 °C. Beads were washed with a buffer (20 mm Tris/HCl, pH 7.5, 7.5150 mm KCl, 2 mm MgCl_2_, 5 mm β‐mercaptoethanol, 50 mm imidazole, 0.5% Triton X‐100, 5% glycerol) on an end‐to‐end rotator for 10 min each at 4 °C. His‐tagged HuR was eluted using various elution buffers containing increasing concentrations of imidazole, each incubated for 15 min at 4 °C with rotation. The final purified protein was aliquoted and stored at −80 °C [11, 21].
Statistical analysis
We used GraphPad Prism 5.00 (Graph Pad, San Diego, CA, USA) to analyze the data plots from experiments conducted in triplicate unless otherwise specified. Student's t‐test was used to calculate P‐values, with significance defined as P < 0.05. Error bars indicate mean ± SD [11, 21].
Results
miR‐122 induces export of miR‐146a from RAW264.7 macrophage cells
Our previous work documented that amino acid starvation‐related stress or high‐lipid exposure induces hepatic cells to secrete EVs containing miR‐122 as a protective mechanism against stress‐induced cell death [25, 36]. EV‐mediated miR‐122 transfer leads to upregulated cytokine expression in miR‐122‐EV‐recipient macrophages or tissue‐resident Kupffer cells, activating them to produce inflammatory cytokines in the high‐lipid‐exposed murine liver [25]. The activation of macrophages is also synchronized with the export of specific miRNAs, a process regulated by the stress response miRNA‐binding protein HuR [26].
This supports the idea that hepatic EVs containing miR‐122 can activate recipient macrophages to release EVs with specific miRNAs (Fig. 1A). Following hepatic EV treatment, we removed the culture medium, washed the cells, and then exposed them to LPS for 4 h. We observed higher levels of miR‐146a in EVs secreted by RAW 264 cells treated with miR‐122‐containing EVs. The increased export of miR‐146a and miR‐122 by these cells after 4 h of LPS treatment was accompanied by reduced cellular levels of these miRNAs. Conversely, the cellular levels of let‐7a remained largely unchanged, although its association with EVs increased after LPS exposure (Fig. 1B,C). This suggests that miRNA export may differentially affect cellular miRNA pools. Additionally, the export of these miRNAs was linked to elevated secretion of inflammatory cytokines IL‐1β and TNF‐α in LPS‐treated cells, indicating that RAW 264.7 cells become more active after LPS exposure (Fig. 1D). Cellular levels of miR‐146a have previously been observed to be downregulated during the initial phase of macrophage activation, followed by an elevation at 24 h of LPS exposure [29]. We propose that the export of miR‐146a from activated macrophage cells accounts for the enhanced expression of inflammatory cytokines in LPS‐treated cells. We also posit that the miR‐146a export is regulated by another miRNA, whose presence in activated macrophages causes the upregulation in EV‐mediated miR‐146a export, which is otherwise not a suitable substrate for the HuR‐mediated miRNA export process [30]. Given that miR‐122 promotes inflammation and miRNA export in macrophages, we suspected that the EV‐derived hepatic miR‐122 could enhance the export of miR‐146a alongside sets of other miRNAs by activating macrophage cells to drive synchronized upregulation of inflammatory cytokines coupled with export of miR‐122‐responsive miRNAs.
*Export of miR‐146a from RAW264.7 cells is controlled by miR‐122. (A) Export of miRNAs from macrophage cells receiving the hepatic miRNAs via EV‐mediated delivery. EVs from Huh7 cells, where miR‐122 is the predominant cargo packaged in the hepatic EVs, were used to treat naïve RAW264.7 cells for 24 h, washed, and then activated by LPS (10 ng·mL−1) for 4 h in EV‐free culture medium. EVs were isolated from treated RAW264.7 cells. Cellular and EV miRNA levels, as well as cellular cytokine mRNAs, were analyzed. (B, C) Levels of EV‐associated (B), and cellular (C) miRNAs from Huh7 EV‐pre‐treated and LPS‐activated RAW264.7 cells and secreted EVs. (for B panel; n = 4, *P = 0.0341, **P = 0.0036 (miR‐146a), **P = 0.0034 (let‐7a); for C panel: n = 4, **P = 0.0088, ***P = <0.0001, *P = 0.0475). (D) Levels of cellular cytokine mRNAs TNF‐α and IL‐1β from LPS‐activated RAW264.7 cells, pretreated with hepatic cell‐derived EVs (n = 4, ***P = 0.0002, **P = 0.0031). (E, F) Levels of cellular (E), and secreted EV‐associated (F) miR‐122 in RAW264.7 cells transfected with increasing concentration of miR‐122 expression plasmid pmiR‐122. Total RNA from cellular lysate was analyzed to quantify the level of miR‐122 by qRT‐PCR, and the dataset without the p‐miR‐122 expression was taken as a unit (n ≥ 3, NS: P = 0.0984, **P = 0.0017 (100 ng), **P = 0.0015 (4000 ng) for panel E; and *P = 0.0215 (100 ng) and *P = 0.0213 (1000 ng) for panel (F). The copy number per cell and per EV (±SD) i plotted and calculated from the miR‐122 standard curve vs. Ct value, as previously described [46]. (G, H) A cooperative effect of miR‐146a packaging occurs during EV‐mediated export. Cellular (G) and secreted EV‐associated (H) miR‐146a levels in cells transfected with increasing miR‐122 expressing plasmid pmiR‐122 were measured. Total RNA from cellular lysate was analyzed to quantify the level of miR‐146a by qRT‐PCR, and the dataset without the pmiR‐122 expression was taken as a unit (for G, n ≥ 4, **P = 0.0095, NS: P = 0.2077(1000ng), NS; P = 0.7036 (4000 ng); For H; n ≥ 5, *P = 0.0105 (100 ng), *P = 0.0328 (1000 ng), NS; P = 0.2729, and unpaired t‐test **P = 0.0022. (I, J) Effect of miR‐122 expression on cellular level of cytokine mRNAs. The level of TNF‐α mRNA showed an increase with cellular miR‐122 level as quantified by qRT‐PCR (n ≥ 4, **P = 0.0045 (100 ng), **P = 0.0096 (1000 ng), ****P = <0.0001 and unpaired t‐test ***P = 0.0008) (I). Levels of IL‐10 mRNA were also analyzed from the same set of samples (n ≥ 3, *P = 0.0324 (100 ng), *P = 0.06369 (1000 ng), **P = 0.0089) (J). The dataset without pmiR‐122 expression was used as a unit for analysis of cytokine mRNAs. (K) miR‐122 expression caused upregulation of inflammatory pathways. P38 MAPK and ERK1/2 activation was evident in western blots done with Phopho‐p38 and phosphor ERK1/2 specific antibodies and compared by western blots between the pCI‐neo transfected control set and the experimental set ectopically expressed miR‐122 (at the level of 1000 ng pmiR‐122 plasmid used to transfect 8 × 106 cells); the condition that showed cooperative miRNA packaging and export. Data information: In all the experimental data, error bars are represented as mean with SD, ns, nonsignificant, *P < 0.05, **P < 0.01, ***P < 0.001, ***P < 0.0001, respectively. P‐values were calculated by a two‐tailed paired t‐test in most of the experiments unless mentioned otherwise. Relative levels of cellular miRNAs were normalized with U6 snRNA level by the 2−ΔΔCt method. The fold change of miRNA was calculated by the 2−ΔCt method. Positions of molecular weight markers are marked and shown in the respective western blots. Normalization was done against the total EV content used for RNA isolation to quantify EV‐associated miRNAs. Relative levels of cellular mRNAs were normalized with GAPDH mRNA levels by the 2−ΔΔCt method.
To study the miR‐122‐controlled export of miR‐146a, we transfected RAW264.7 cells with miR‐122 expression plasmids to induce ectopic expression of miR‐122 in these cells. We documented an increase in its expression alongside increased EV‐mediated export of miR‐146a derived from miR‐122‐expressing RAW264.7 cells in a dose‐dependent manner proportional to the rise in cellular miR‐122 levels (Fig. 1E,F). Interestingly, the EV‐associated miR‐146a also increases with higher miR‐122 expression until a decrease in EV‐miR‐146a levels occurs when cellular miR‐122 content reaches a possible threshold level. The cellular miR‐146a levels changed reciprocally, as expected. However, changes in cellular miR‐146a content were modest until the export of miR‐146a was reduced at the highest levels of cellular miR‐122 expression, accompanied by an increase in cellular miR‐146a content (Fig. 1G,H). These data suggest a biphasic effect of miR‐122 on miR‐146a expression. While low miR‐122 levels promoted miR‐146a export and restricted cellular miRNA content, high levels of cellular miR‐122 acted as a competitive inhibitor of miR‐146a export from RAW264.7 cells. The mechanism underlying miR‐146a export inhibition at higher miR‐122 levels remains unclear. It may serve as a safeguard against inflammatory overactivation, as miR‐146a protects macrophages during prolonged immune activation by upregulating secondary miRNA biogenesis [29]. Repression of inflammatory cytokines by miR‐146a has been reported previously [30]. Interestingly, in RAW264.7 macrophages, at the highest miR‐122 level, miR‐146a export decreases, whereas TNF‐α production increases. This indicates that miR‐146a, which is not properly exported, accumulates in a nonfunctional state when miR‐122 levels are high, thereby increasing TNF‐α production due to impaired miR‐146a‐mediated translational repression. Previous studies have reported inactivation of miR‐146a in C6 and primary astroglial cells exposed to amyloid protein, along with its accumulation in RNA‐processing bodies and enhanced expression of inflammatory cytokines [37, 38]. We assume that a similar mechanism may account for the nonfunctionality of export‐retarded miR‐146a, happening with excess miR‐122.
Consistent with our previous observation of EV‐associated miR‐122 treatment‐induced expression of inflammatory cytokines in macrophages [25, 36], we documented an enhancement of TNF‐α in miR‐122‐expressing cells with a marginal effect on IL‐10 anti‐inflammatory cytokine expression (Fig. 1I,J). At a plasmid concentration of 1000 ng used to transfect 8 × 10^^6^ cells (approximately 5300 copies of miR‐122/cell), miR‐122‐induced miR‐146a export was observed. We assessed immune activation by measuring signaling components in miR‐122‐expressing cells. We observed that miR‐122 activates the p‐p38 MAPK and p‐ERK1/2 pathways in miR‐122‐expressing RAW264.7 cells (Fig. 1K), but without a significant effect on Ago2, HuR, or c‐PARP levels, which signifies no substantial change in stress or cell death with pmiR‐122 transfection.
In LPS‐activated macrophages, we observed miR‐146a induction occurring during the late phase of activation (24 h), which was associated with the miR‐146a‐dependent biogenesis of ‘secondary’ miRNAs from respective pre‐miRNAs. This ‘cooperative biogenesis’ of miRNAs results in increased mature miRNA levels for secondary miRNAs regulated by miR‐146a and a concomitant decrease in the respective pre‐miRNA levels [39]. Interestingly, miR‐122 expression, which enhances miR‐146a expression and export, does not induce coordinated biogenesis of ‘secondary’ miRNAs, as there are no significant changes in the mature and pre‐miRNA levels for ‘secondary’ miR‐125b or miR‐143‐3p (Fig. S1), clearly showing the coordinated miRNA export phase is separate from that of coordinated biogenesis reported earlier for the late phase of macrophage activation [29].
Cooperative entry of miRNA into endosomes is dependent on HuR
miRNAs need to be transported to endosomes for subsequent EV‐mediated export [11]. We recently developed an in vitro miRNA import assay using isolated endosomes to study miRNA entry into endosomes. We documented concentration‐dependent entry of single‐stranded miRNAs into the endosomal lumen, which becomes resistant to RNase digestion after the import reaction [21]. To test cooperativity in endosomal miRNA entry, we conducted an in vitro import assay for miR‐146a in the presence of increasing concentrations of miR‐122, using endosomes isolated from C6 glioblastoma cells [21]. Interestingly, in line with our in vivo observations, we found a concentration‐dependent positive effect of miR‐122 on miR‐146a entry into endosomes (Fig. 2A,B). These data are consistent with the in vivo results we obtained with pmiR‐122‐transfected RAW264.7 cells for miR‐146a, in which the presence of miR‐122 at a low ‘catalytic’ amount positively affected miR‐146a export.
*miR‐122 promotes cooperative packaging of miRNAs, including miR‐146a, into endosomes. (A) Scheme of the in vitro endosome targeting assay of miRNAs. The diagram depicts cooperative miRNA packaging in an in vitro assay using enriched endosomes isolated from C6 rat glioma cells. Under different conditions, as required by specific experiments, synthetic single‐stranded miRNAs such as ss miR‐122, ss‐miR‐146a, ss‐miR‐155, or a pool of small RNAs isolated from RAW264.7 cells were used as cargo miRNA to observe the cooperation and competition between multiple miRNAs for the packaging into endosomes following previously published procedures [21]. (B) Cooperative effect of endosomal packaging of two species of miRNAs. Synthetic ss‐miR‐146a (10 nm) was added as the cargo and incubated for 30 min with increasing amounts of ss‐miR‐122 (0, 0.1, 1, 2, 4 nm). Excess nonimported RNA was removed by RNase treatment, followed by re‐isolation of endosomes. qRT‐PCR quantified the packaged miRNAs. The dataset at 0 nm ss‐miR‐122 was used as the unit. n ≥ 3, *P = 0.0273 (0.1 nm), *P = 0.0227 (1 nm), *P = 0.0177 (2 nm), *P = 0.0184 (4 nm). (C) With endosomes isolated from C6 cells, 1000 ng·mL−1 of small RNA pool isolated by miRVana kit (Thermo Scientific) was added with an increasing concentration of synthetic single‐stranded (ss) miR‐122. miRNA packaging reaction was carried out for 30 min followed by an RNase protection and vesicle re‐isolation (details in the Methods section). qRT‐PCR measured packaged levels of various miRNAs, and the value of individual small RNA in the presence of 0 nm ss‐miR‐122 was considered a unit. Quantitative measurement for endosome targeted miR‐21 (left) and let7a (right) miRNA [miR‐21; n = 3, NS; P = 0.2671, **P = 0.0024, *P = 0.0414; let‐7a; n ≥ 3, *P = 0.0239 (2 nm), **P = 0.0049, *P = 0.0168 (20 nm) are shown]. (D) Influence of ss‐miR‐122 (4 nm) on endosomal packaging of 10 nm of ss‐miR‐155a. Excess reactant RNA was removed by RNase reaction and vesicle re‐isolation. The packaged miRNA was quantified by qRT‐PCR and the dataset with 0 nm ss‐miR‐122 was considered as a unit (n = 4, **P = 0.0061). (E) Endosomal packaging of ss‐miR‐122 (10 nm) was studied in the presence of ss‐miR‐146a or ss‐miR‐155 (4 nm). After 30 min of incubation, excess unpackaged RNA was eliminated by RNase reaction and vesicle re‐isolation. The internalized miR‐122 was quantified by qRT‐PCR and the dataset with 0 nm ss‐miR‐146a (left panel) or 0 nm ss‐miR‐155 (right panel) was considered as unit (left panel n = 6, **P = 0.0054; right panel n = 5, ***P = 0.0005). (F) Cumulative cooperative effect of ss‐miR‐122 (0.1 nm) and recombinant HuR (rHuR, 50 nm) on endosomal packaging of ss‐miR‐146a (10 nm). The reaction was carried out for 30 min; excess unpackaged RNA was depleted by RNase reaction and vesicle re‐isolation. QRT‐PCR quantified the packaged miRNA, and the dataset excluding ss‐miR‐122 and rHuR was used as unit. Western blot shows the level of HuR from the samples after the vesicles were re‐isolated. n ≥ 3, NS; P = 0.0521, *P = 0.0348 (no miR‐122; with HuR), *P = 0.0396 (with miR‐122 and HuR); unpaired t‐test P = 0.0369. Data information: In all the experimental data, error bars are represented as mean with SD, ns, nonsignificant, *P < 0.05, **P < 0.01, **P < 0.001, respectively. P‐values were calculated by a two‐tailed paired t‐test in most of the experiments unless mentioned otherwise. The fold change of miRNA was calculated by 2−ΔCt method. Endosomal miRNA level normalization was done against the total endosomal content used for the assay to quantify endosome‐associated miRNAs. Positions of molecular weight markers are shown for the respective western blots.
To test whether miR‐122's effect remains exclusive to miR‐146a, we incubated the small RNA pool isolated from C6 cells with increasing concentrations of synthetic single‐stranded (ss) miR‐122. We observed a dose‐dependent increase in the import of let‐7a and miR‐21 by miR‐122. Interestingly, while the 10 mm concentration of miR‐122 enhances the entry of miR‐21 and let‐7a into endosomes, a 20 mm concentration of miR‐122 substantially reduces the import level of let‐7a and begins to show a decrease in miR‐21 endosomal import levels as well (Fig. 2C). When synthetic miR‐155 was used as a secondary substrate at a 4 nm miR‐122 concentration, cooperative entry of miR‐155 was also observed in an endosomal targeting assay in vitro (Fig. 2D)‐suggesting cooperative effect of miR‐122 on endosomal loading of other miRNAs apart from miR‐146a.
While catalytic amount of miR‐122 promotes export of other miRNAs substates, at higher concentrations, miR‐122 exerted an inhibitory competitive effect (Figs 2C, 1H). Interestingly, with miR‐146a or miR‐155 used at lower concentrations, both miRNAs inhibited the endosomal entry of synthetic ssmiR‐122 (Fig. 2E). These data suggest that the cooperative effect of one miRNA on another is unidirectional and specific. At higher concentrations, the same miRNAs may act as competitive inhibitors. In contrast, at ‘catalytic’ concentrations, the cooperativity of miRNA entry into endosomes, driven by a primary catalytic miRNA such as miR‐122, has been documented that enables subsequent EV‐mediated export of a set of coregulated miRNAs.
HuR is known to regulate the miRNA export process by reversibly binding of the miRNAs to facilitate the endosomal miRNA entry [11, 21]. To test the effect of HuR on the cooperative entry of miRNAs into endosomes, we conducted an endosomal import assay for miR‐146a in the presence of HuR and ssmiR‐122 and observed enhanced endosomal import of miR‐146a when recombinant HuR and miR‐122 were added separately. The import of miR‐146a in the presence of miR‐122 was significantly improved with recombinant HuR (Fig. 2F), suggesting an additive effect of miR‐122 and HuR on miR‐146a entry into endosomes.
To confirm that endosomal compartmentalization of miRNAs occurs cooperatively in the in vivo context and that EV‐associated miRNA export is proportional to this cooperative endosomal targeting, we expressed miR‐122 in RAW264.7 cells alone or with HA‐HuR to evaluate the effects on subcellular miR‐122 and miR‐146a content, as well as changes in their EV association. With the expression of miR‐122, there was an increase in EV content released by pmiR‐122‐expressing cells, which was significantly enhanced with the co‐expression of HA‐HuR in RAW264.7 cells (Fig. S2A–D). This may explain the activation state of RAW264.7 cells, which is associated with miR‐122 expression and improved miRNA and EV export. We performed subcellular fractionation of cell lysates on an Optiprep density gradient to separate endosomes and the ER, and we quantified the amounts of ER and endosome‐associated miRNAs obtained from pCI‐neo control, pmiR‐122, and pmiR‐122 plus pHA‐HuR cotransfected cells (Fig. S3A,B) [21, 40]. Upon transfection with pmiR‐122 and pHA‐HuR, we observed increased levels of miR‐122 in EVs and endosomes (Fig. S3C). At the same time, a clear enrichment of miR‐146a content in EVs and endosomes was also noted (Fig. S3D). Thus, both in vitro and in vivo, miR‐122 and HA‐HuR cooperatively enhance the endosomal targeting of miR‐146a. This explains the enhanced export of miR‐146a via EVs from cells expressing low levels of miR‐122.
High‐affinity miR‐122 enhances the binding of HuR with low‐affinity substrate miR‐146a
The stress response protein HuR is a miRNA binder and is necessary and sufficient for miR‐122's import into endosomes and EV‐mediated export in hepatic cells under stress [11]. HuR is also required for the export of miRNAs such as let‐7a and miR‐155 from LPS‐activated RAW264.7 macrophage cells [26]. These miRNAs are found to be regulated cooperatively by miR‐122a (Fig. 2C,D), while cooperative entry of miR‐146a into endosomes is also facilitated by HuR (Fig. 2F).
HuR binds with its miRNA substrates with differential affinity, possibly determined by the presence or absence of AU‐rich sequences on the RNA substrates. We used HA‐HuR immobilized on Protein‐G agarose beads, which were incubated with a mix of ssmiR‐122 and ssmiR‐146a. To identify the cooperative effect of miR‐122 on miR‐146a HuR binding, we incubated 10 nm miR‐122 with HuR immobilized beads in the presence of increasing concentrations of miR‐146a. For a fixed amount of miR‐122 as substrate, the increasing concentration of miR‐146a caused a concentration‐dependent decrease in miR‐122 binding to HuR. Conversely, miR‐146a binding remained low until a threshold concentration of miR‐146a caused a jump in HuR‐miR‐146a binding at 4 nm (Fig. 3A,B). With a fixed amount of miR‐146a used in a binding assay, increasing the miR‐122 concentration resulted in a sharp rise in miR‐146a binding with 1 and 2 nm of miR‐122 present. In contrast, a drop in miR‐146a binding to HuR was noted with 4 nm of miR‐122 (Fig. 3C). These data suggest that cooperative binding of miR‐146a to HuR occurs at lower concentrations of miR‐122.
*Cooperative binding of low‐affinity miR‐146a to HuR in the presence of high‐affinity miR‐122. (A) Scheme of the experiments. The HA‐HuR expression vector was transfected into HEK293 cells, and from the cellular lysate, HA‐HuR was immunoprecipitated and immobilized on HA‐antibody‐bound Protein‐G agarose beads. Equal amounts of immobilized HA‐HuR were incubated in a HuR‐binding assay buffer (details in the Methods section) with substrate synthetic single‐stranded miRNA for 15 min at room temperature. After the reaction, beads were pelleted down and washed to remove unbound components. The beads were divided equally to analyze miRNAs and proteins from these samples. (B) Suggested as assay 2 in panel A; synthetic ss‐miR‐122 (10 nm) was added in the immobilized HA‐HuR reaction system along with varying amounts of synthetic ss‐miR‐146a (0, 1, 2, 4 nm). After incubation, the beads were thoroughly washed to remove excess or nonspecific RNAs in the wash buffer, and the samples were then divided equally for RNA and protein analysis. The HuR‐associated miR‐122 and miR‐146a levels were analyzed by qRT‐PCR, using the 0 nm ss‐miR‐146a dataset as the unit (n = 2). The amount of HuR isolated as immunoprecipitate was used for normalization. (C) Suggested as assay 1 in panel A; synthetic ss‐miR‐146a (10 nm) was added in the immobilized HA‐HuR reaction system along with varying amounts of secondary miRNA species synthetic ss‐miR‐122 (0, 1, 2, 4 nm), and the HuR association reaction was carried out. After the reaction, the beads were thoroughly washed to remove excess reactants, and then, the samples were divided equally for RNA and protein analysis. qRT‐PCR analyzed the HuR‐associated miR‐146a levels by considering the dataset of 0 nm ss‐miR‐122 as a unit (n = 3, *P = 0.0186, **P = 0.0096, NS; P = 0.0701) (lower panel). The amount of HuR isolated as immunoprecipitate was used for normalization (upper panel). Data information: In all the experimental data, error bars are represented as mean with SD, ns, nonsignificant, *P < 0.05, **P < 0.01, **P < 0.001, respectively. P‐values were calculated by a two‐tailed paired t‐test in most of the experiments unless mentioned otherwise. The fold change of miRNA was calculated by the 2−ΔCt method.
miR‐122‐induced miRNA export regulates the inflammatory responses
The consequences of this cooperative export of miRNAs by macrophages are not well understood. We assume that the export of miRNAs from 4 h LPS‐activated or miR‐122‐expressing RAW264.7 cells is essential for activating macrophages. To test whether miRNA export may be linked to the activation of RAW264.7 cells, we block the export of miRNAs using GW4869 in cells expressing miR‐122 in an inducible manner [11]. Inducible expression of miR‐122 in RAW264.7 cells has been achieved with doxycycline [29] (Fig. 4A). In miR‐122‐expressing cells, blocking EV‐mediated export repressed TNF‐α and IL‐1β mRNA levels, which were partially rescued by anti‐miR‐146a treatment (Fig. 4B). This suggests the importance of miR‐122 and miR‐122‐regulated miR‐146a export in inflammatory response. Previous data from our lab established that miR‐146a mediates an anti‐inflammatory response, with miR‐146a upregulation linked to downregulation of inflammatory cytokines in RAW264.7 macrophages [30]. Thus, the current data are consistent with previous observations, as blocking miR‐146a activity rescues IL‐1β and TNF‐α expression in cells that are blocked from extracellular miRNA export by GW4869 and that express miR‐122. However, the effect was more pronounced on IL‐1β than on TNF‐α (Fig. 4B).
*Cooperative export of miR‐146a is essential for proinflammatory responses in cells expressing miR‐122. (A) Scheme of the experiment. RAW264.7 cells stably expressing Tet‐On expression cassette were transfected with doxycycline inducible‐miR122 expression plasmid (ipMIR‐122) for 24 h. These cells were subjected to EV secretion blocker GW4869 (10 μm, 16 h in presence or absence of miR‐146a inhibitory anti‐miR‐146a oligos, 30 nm, 24 h) to test its effects on cytokine expression. miR‐122 expression was induced with doxycycline (400 ng·mL−1, 24 h). The cellular levels of key cytokine mRNAs were measured. (B) The cellular levels of TNF‐α and IL‐1β mRNAs under the different treatment conditions were analyzed by qRT‐PCR (TNF‐α; n = 4, **P = 0.0056, NS; P = 0.2063; IL‐1β; n = 4, **P = 0.0053, *P = 0.0470; and unpaired t‐test NS; P = 0.1438 for TNFα and ***P = 0.0005 for IL‐1β). (C) Scheme of the experiment done with Leishmania donovani infection of RAW264.7 macrophages. Cells stably expressing Tet‐On expression cassette were transfected with doxycycline inducible‐miR122 vector (ipmiR‐122). These cells were subjected to miR‐146a inhibitor (anti‐miR‐146a oligos, 30 nm, 24 h) and/or EV secretion blocker‐ GW4869 treatment (10 μm, 16 h). miR‐122 expression was induced with doxycycline (400 ng·mL−1, 24 h), which was followed by infecting the cells with Leishmania donovani (Ld) promastigotes (1:10 ratio of macrophage to Ld) for 6 h; noninternalized Ld were washed away. Then, the cells were allowed to recover for another 12 h post‐infection as done previously [30]. The cellular levels of a key cytokine mRNA and pathogen‐specific alpha ‐amastin mRNA were measured. (D) The cellular levels of TNF‐α and IL‐1β cytokine mRNAs, along with α‐amastin levels, were analyzed by qRT‐PCR to determine the levels of inflammatory condition and Ld infection under different treatment and infection conditions. For TNF‐α, n = 4, ***P = 0.0010, NS; P = 0.5847, *P = 0.0158; for IL‐1β, n = 4, **P = 0.0074 (with GW4869; no anti‐miR‐146a), NS; P = 0.6831, **P = 0.0012 (with GW4869 and anti‐miR‐146a); for α‐amastin, n = 4, ***P = 0.0001, *P = 0.0191 (with anti‐miR‐146a, no GW4869), *P = 0.0372 (with anti‐miR‐146a and GW4869); and unpaired t‐test **P = 0.0088 for TNFα, *P = 0.0103 for IL‐1β and **P = 0.0022 for α‐amastin. Data information: In all the experimental data, error bars are represented as mean with SD, ns, nonsignificant, *P < 0.05, **P < 0.01, **P < 0.001, respectively. P‐values were calculated by a two‐tailed paired t‐test in most of the experiments unless mentioned otherwise. Relative levels of mRNAs were normalized with GAPDH mRNA levels by 2–ΔΔCt method.
miR‐146a plays a crucial role in the anti‐inflammatory response. The upregulation of miR‐146a is essential for Leishmania donovani infection of macrophages [30]. The miR‐146a‐containing extracellular vesicles (EVs) derived from infected macrophages are also transferred to naïve macrophages, polarizing them to an M2‐like phenotype by increasing miR‐146a content in the recipient cells, thereby downregulating inflammatory cytokines [30]. We documented an increased level of infection, as measured by the internalization of the pathogen‐specific gene α‐amastin, in cells inducibly expressing miR‐122 but treated with GW4869, which blocks miRNA export and is known to reduce inflammatory cytokine expression in RAW264.7 cells [26] (Fig. 4C,D). We observed no change in cytokine expression following anti‐miR‐146a administration. Still, a substantial reduction in cytokine expression accompanied by upregulated α‐amastin expression, suggesting higher infection at lower inflammatory cytokine levels with GW4869 (Fig. 4D). These data indicate possible involvement of separate miRNAs, other than miR‐146a, in the regulation of Ld infection and related cytokine suppression in Ld‐infected cells. Overall, these results are consistent with the hypothesis that miR‐122‐induced coordinated export of miRNAs may trigger inflammatory responses and that this effect can be reversed by inhibiting the export of the respective miRNAs to prevent miR‐122‐induced inflammation. Thus, coordinated miRNA export can be targeted to restrict inflammatory responses in mammalian macrophages.
Discussion
Our previous observations suggest that LPS induces the uncoupling of miRNAs from phosphorylated Ago2, which leads to the accumulation of Ago2‐free from miRNAs [41]. The fate of Ago‐free miRNAs has not been investigated, but they are expected to be exported by activated macrophages [26, 30]. The coordinated export may indicate how the balanced expression of Ago‐uncoupled miRNAs can serve as substrates for EV‐mediated export. However, the relationship between cooperative export of miRNAs and Ago2's miRNA‐unbinding remains unknown.
The cooperative import of miRNAs into endosomes is linked to the co‐export of miRNAs documented in RAW 264.7 cells. The miRNA‐binding protein HuR is essential for the cooperative import of miRNAs into endosomes and the EV‐mediated co‐export of miRNAs. HuR's binding with high‐affinity miR‐122 enhances the binding of low‐affinity miRNA substrates to HuR, facilitating the co‐entry of both miRNAs into endosomes. We documented earlier that the RRMIII domain of HuR is crucial for miRNA export and miRNA‐binding activity [11]. At the same time, the hinge region of HuR is responsible for HuR ubiquitination and EV‐mediated miRNA export [11]. We suggest that HuR's binding to high‐affinity miRNAs could cause a conformational change that improves its ability to bind with secondary low‐affinity miRNAs, potentially via the other two RRMs of HuR. Alternatively, this may be due to faster oligomerization, which promotes entrapment of Ago2‐free miRNAs within HuR oligomers [42]. Oligomerization of HuR has been studied previously and has been found to be essential for mRNA stabilization [43] and miRNA inactivation [42]. The HuR‐miRNA complex subsequently activates the RalA GTPase and causes selective, but cooperative, import of miRNAs into endosomes [21]. We have shown earlier that the HuR‐miRNA complex can also transfer miRNAs to the syntaxin protein STX5, which synergizes with HuR to facilitate EV‐mediated miRNA export [40]. It is unclear how STX5 or RalA may affect cooperativity of miRNA import into endosomes [40].
The cooperativity of miRNA binding with HuR can explain the cooperative miRNA export we have observed in macrophages. We posit that in the presence of a catalytic amount of miR‐122 (the high‐affinity binder), the HuR‐miR‐122 complex forms, allowing the formation of the miR‐146a‐HuR complex (Fig. 5A). HuR forms oligomers with the RNA substrates. HuR oligomerization on AU‐rich sequences of target mRNAs is required for miRNA replacement from target mRNAs and miRNA derepression [44]. Thus, HuR oligomerization in the presence of the high‐affinity substrate miR‐122 may increase the affinity of oligomeric HuR for the low‐affinity substrate miR‐146a, as the latter gets trapped in a co‐oligomer of HuR and miR‐122 (Fig. 5A). At higher concentrations, miR‐122 competitively inhibits miR‐146a export and acts as an antagonist. This agonist‐to‐antagonist functional switch may be a unique strategy to balance miR‐146a levels via miR‐122, but the mechanism remains unknown. We posit that miR‐122 binding alters the affinity of the 2nd miRNA‐binding site for HuR, thereby making miR‐146a a better substrate than miR‐122; thus, only at higher miR‐122 concentrations can miR‐122 compete with miR‐146a for the 2^nd^ miRNA‐binding site of HuR, explaining the agonist‐to‐antagonist functional switching of miR‐122. In the current model of HuR‐mediated miRNA export, double‐stranded miRNAs may serve as substrates for mRNA export. However, they must dissociate from the passenger strand, bind HuR, and be transported to endosomes [21].
Biphasic cooperative regulation of miRNAs in mammalian macrophages. (A) Possible model of cooperative binding of HuR with miR‐146a in the presence of miR‐122. High‐affinity miR‐122 promotes the low binder miR‐146a binding with HuR, where HuR oligomerization, favored by miR‐122, may assist the miR‐146a binding or entrapment within HuR oligomers. At higher concentrations, abundant miR‐122 inhibits miR‐146a binding in an antagonistic manner. (B) The biphasic regulation of miRNAs is observed in mammalian cells. Induction of cooperative export induced by exogenous miR‐122 or specific endogenous miRNA (miR‐A) caused cooperative export of secondary miRNAs regulating specific cytokine and related mRNAs to ensure robust and cooperative expression during the early phase of macrophage activation, essential for pro‐inflammatory cytokine expression [29]. The late anti‐inflammatory reactions are caused by a specific member of the miRNAs induced by late‐phase‐associated miRNA(s) (miR‐146a; miR‐X) that cause coordinated biogenesis of other miRNAs to ensure repression of cytokine expression in the late phase or overly activated macrophage to protect the macrophage from apoptotic death as part of the essential LPS‐tolerance mechanism. (C) The published data suggest biphasic changes in miR‐146a expression levels in activated macrophages [29]. The secondary miR‐143‐5p follows a late response expression pattern that accounts for the bell‐shaped TNF‐α expression pattern [29]. This model shows how cooperative export and biogenesis contribute to immune activation regulation in macrophage cells during LPS‐induced activation. (D) Dose–response data of miR‐122 and miR‐146a suggest that miR‐122, at low concentration, contributes to the cooperative export of miR‐146a, while, at higher concentration, competitively prevents the export of miR‐146a. Thus, miR‐122 may contribute to the cooperative biogenesis step of miRNAs induced by accumulated miR‐146a to optimize the immune response. (E) A graphical summary of the macrophage activation process controlled by cooperative export of miR‐146a by miR‐122. Expression or uptake of miR‐122 in macrophages induced the export of miR‐146a (and other coregulated miRNAs), a repressive miRNA that inhibits the expression of inflammatory cytokines, thereby activating macrophages.
The coordinated miRNA regulation and export observed in the early phase of macrophage activation ensure the systematic inactivation and export of miRNAs from macrophage cells. This allows cells to respond to activation signals while simultaneously permitting the coordinated expression of cytokine‐encoding mRNAs that are repressed by miRNAs in activated macrophages [41]. Thus, the EV‐mediated coordinated export, in turn, regulates specific sets of miRNAs and their target genes simultaneously.
During the late phase of macrophage activation, coordinated re‐repression of miRNA targets must be ensured to prevent damage from unregulated and excess cytokine production damaging to the macrophage [29, 39]. The coordinated biogenesis of secondary miRNAs is achieved by increased expression of primary miRNAs, such as miR‐146a, which induces the biogenesis of sets of secondary miRNAs [29]. miR‐146a in RAW264.7 cells shows a biphasic expression pattern. Cooperative export‐mediated downregulation of miR‐146a during the early phase of activation by miR‐122 or LPS induces a surge in cytokine expression. However, with an increasing amount of miR‐122 accumulating, it gradually acts competitively to block the export of miR‐146a, allowing the accumulation of miR‐146a, possibly resulting in coordinated biogenesis of secondary miRNAs occurring during the late phase of macrophage activation, as it is also noted in LPS‐treated macrophages (Fig. 5A,B). Therefore, miR‐146a is a key regulatory miRNA, whose expression is controlled by coordinated export, ensuring regulated cytokine production. At a later stage, the accumulation of miR‐146a drives coordinated miRNA biogenesis, suppressing the expression of many cytokines during the late activation phase. The importance of both the coordinated regulation of miR‐146a export and miR‐146a‐driven miRNA biogenesis in mammalian macrophage cells has clear physiological implications in inflammatory responses and tissue‐specific inflammation regulation in the liver under nonalcoholic steatohepatitis (NASH) or in degenerating brains on exposure to amyloid proteins (Fig. 5C,D).
We have identified miR‐122 as the central hepatic cell‐derived miRNA responsible for inflammation in murine livers exposed to a high‐fat diet. The miR‐122‐containing EVs released by hepatic cells upon exposure to high lipids are taken up by Kupffer cells, where they induce inflammatory cytokines. We posit that miR‐122, upon entering macrophages, induces the coordinated export of the anti‐inflammatory miR‐146a as a feedback mechanism that delivers it to hepatic cells, thereby reducing miR‐122 expression in those cells (Fig. 5E). The reduction in miR‐122 expression by miR‐146a and miR‐146a‐containing EVs has been documented in other contexts, supporting our hypothesis of reciprocal regulation occurring in fatty liver [30]. Thus, macrophage‐hepatocyte crosstalk via EVs, with coordinated miRNA export, may be essential in controlling metaflammation‐related liver damage.
The same miRNA‐146a, while regulating it in the presence of miR‐122 in noninfected macrophages, seems not to affect cytokine repression in Ld‐infected cells. This may be because the cytokine repression is mediated by the Ld parasite‐derived GP63 protease, which downregulates cytokine mRNA binding (and the stabilizer) protein HuR in infected cells, thereby reducing cytokine levels. Additionally, HuR works alongside other miRNAs that suppress cytokines [26].
Regarding the possible mechanism of cooperative export of miRNAs, we consider several factors, including the presence of target mRNAs with binding sites for both miRNAs, which may contribute to coordinated miRNA export, as has been reported in the context of cooperative miRNA biogenesis [45]. The importance of miRNA binding to the Ago protein is also reflected in its regulation of EV‐mediated miRNA export, and we posit that modifications of miRNAs, such as uridylation of m6A, may allow the unbinding of miRNAs from Ago2, thereby facilitating their export [22]. How these changes are linked to the cooperative export of miRNAs is an unanswered question. Previous experimental data confirmed the single‐stranded miRNA's entry into endosomes for EV packaging, and the process does not depend on Ago. However, a portion of the HuR‐miRNA complex may remain bound to the endosome membrane and contribute to the background signals we observed at high concentrations of recombinant HuR in the assay. The possibility that non‐EV miRNA export contributes to changes in miR‐146a or other miRNA levels in extracellular content remains unaddressed. It is also possible that miR‐122–induced cooperative export plays a role in altering non‐EV extracellular miRNA content, along with the changes in the Ago2‐miRNA complex present in the extracellular matrix.
Author contributions
SG was involved in data curation, formal analysis, investigation, and methodology. KM was involved in data curation, formal analysis, investigation, writing‐review and editing, conceptualization, methodology, and supervision. SNB was involved in data curation, formal analysis, investigation, writing‐review and editing, conceptualization, funding acquisition, methodology, and supervision.
Supporting information
Fig. S1. Effect of coordinated miR‐146a export by miR‐122 on biogenesis of secondary miRNAs in RAW264.7 cells. Fig. S2. Ectopic expression of miR‐122 and HA‐HuR enhances the EV production in RAW264.7 cells. Fig. S3. Effect of miR‐122 and HA‐HuR expression on the subcellular and EV‐associated miR‐146a levels. Table S1. List of Plasmids and Chemicals. Table S2. Details of miRNA primers used for Taqman‐based quantification. Table S3. List of synthetic single‐stranded RNA and anti‐miR oligonucleotides. Table S4. Details of mRNA specific primers used for SYBR‐Green‐based quantification. Table S5. Details of antibodies used for Western blot (WB), Immunofluorescence (IF), and Immunoprecipitation (IP).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Filipowicz W , Bhattacharyya SN and Sonenberg N (2008) Mechanisms of post‐transcriptional regulation by micro RN As: are the answers in sight? Nat Rev Genet 9, 102–114.18197166 10.1038/nrg 2290 · doi ↗ · pubmed ↗
- 2Bartel DP (2009) Micro RN As: target recognition and regulatory functions. Cell 136, 215–233.19167326 10.1016/j.cell.2009.01.002PMC 3794896 · doi ↗ · pubmed ↗
- 3Gurtan AM and Sharp PA (2013) The role of mi RN As in regulating gene expression networks. J Mol Biol 425, 3582–3600.23500488 10.1016/j.jmb.2013.03.007PMC 3757117 · doi ↗ · pubmed ↗
- 4Pillai RS , Bhattacharyya SN , Artus CG , Zoller T , Cougot N , Basyuk E , Bertrand E and Filipowicz W (2005) Inhibition of translational initiation by Let‐7 Micro RNA in human cells. Science 309, 1573–1576.16081698 10.1126/science.1115079 · doi ↗ · pubmed ↗
- 5Pratt AJ and Mac Rae IJ (2009) The RNA‐induced silencing complex: a versatile gene‐silencing machine. J Biol Chem 284, 17897–17901.19342379 10.1074/jbc.R 900012200 PMC 2709356 · doi ↗ · pubmed ↗
- 6Garcia‐Martin R , Wang G , Brandão BB , Zanotto TM , Shah S , Kumar Patel S , Schilling B and Kahn CR (2022) Micro RNA sequence codes for small extracellular vesicle release and cellular retention. Nature 601, 446–451.34937935 10.1038/s 41586-021-04234-3PMC 9035265 · doi ↗ · pubmed ↗
- 7Groot M and Lee H (2020) Sorting mechanisms for Micro RN As into extracellular vesicles and their associated diseases. Cells 9, 1044.32331346 10.3390/cells 9041044 PMC 7226101 · doi ↗ · pubmed ↗
- 8Valadi H , Ekström K , Bossios A , Sjöstrand M , Lee JJ and Lötvall JO (2007) Exosome‐mediated transfer of m RN As and micro RN As is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9, 654–659.17486113 10.1038/ncb 1596 · doi ↗ · pubmed ↗