Antischistosomal activity of imidazolidine-2,4-dione derivatives
Anna Jaromin, Anna Czopek, Thainá R. Teixeira, Agnieszka Zagórska, Josué de Moraes

TL;DR
Researchers tested new compounds for treating schistosomiasis and found some effective and safe options.
Contribution
Identified imidazolidine-2,4-dione derivatives as a novel scaffold with antischistosomal activity.
Findings
Four imidazolidine-2,4-dione derivatives showed selective antischistosomal activity.
These compounds had low cytotoxicity in mammalian cells.
Isoindole-1,3-dione derivatives were ineffective against the parasite.
Abstract
Schistosomiasis is a neglected tropical disease with limited treatment options and growing concerns over praziquantel resistance. We evaluated a series of 12 synthetic compounds for antiparasitic activity against Schistosoma mansoni adult worms in vitro. Isoindole-1,3-dione derivatives were inactive, whereas four imidazolidine-2,4-dione derivatives displayed selective antischistosomal activity with low cytotoxicity in mammalian cells. These findings highlight imidazolidine-2,4-dione as a promising scaffold for the development of new therapeutic agents against schistosomiasis.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
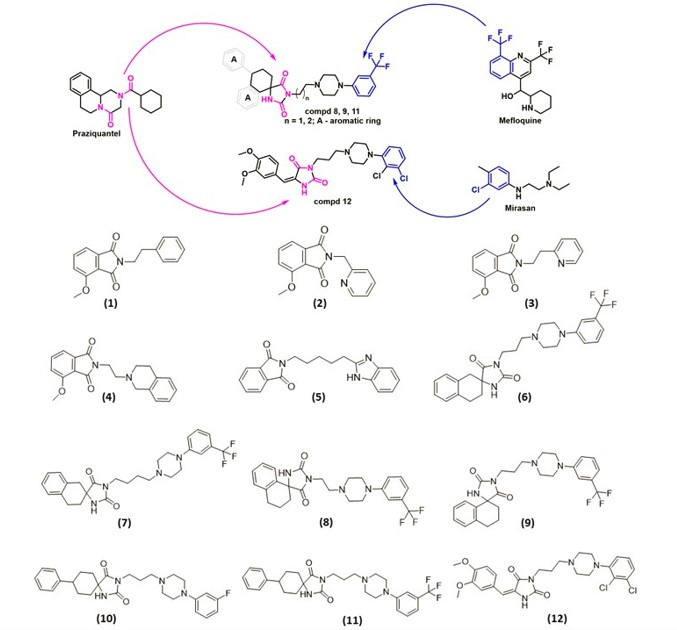 Fig 1
Fig 1| Compound | Vero cells | SI | |
|---|---|---|---|
| 1 | >50 | ND | ND |
| 2 | >50 | ND | ND |
| 3 | >50 | ND | ND |
| 4 | >50 | ND | ND |
| 5 | >50 | ND | ND |
| 6 | >50 | ND | ND |
| 7 | >50 | ND | ND |
| 8 | 18.1 (17.2–22.5) | >200 | >11.05 |
| 9 | 12.6 (9.7–16.4) | >200 | >15.87 |
| 10 | >50 | ND | ND |
| 11 | 19.8 (17.6–23.9) | >200 | >10.1 |
| 12 | 13.3 (10.1–17.3) | >200 | >15.03 |
| PZQ | 0.98 (0.7–1.2) | >200 | >204 |
- —Fundação de Amparo à Pesquisa do Estado de São Paulohttp://dx.doi.org/10.13039/501100001807
- —Coordenação de Aperfeiçoamento de Pessoal de Nível Superiorhttp://dx.doi.org/10.13039/501100002322
- —Conselho Nacional de Desenvolvimento Científico e Tecnológicohttp://dx.doi.org/10.13039/501100003593
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsParasites and Host Interactions · Research on Leishmaniasis Studies · Synthesis of heterocyclic compounds
INTRODUCTION
Schistosomiasis is a neglected tropical disease caused by parasitic flatworms of the genus Schistosoma. It affects approximately 250 million people worldwide, with significant morbidity and socioeconomic burden in endemic regions, particularly in sub-Saharan Africa, South America, and parts of Asia (1). Praziquantel, a synthesized pyrazine isoquinoline derivative, is currently the only widely used drug, and the risk of resistance has prompted the search for new chemical entities (2, 3).
To address these challenges and meet the United Nations Sustainable Development Goals, the World Health Organization has established a 2021–2030 roadmap for the control and elimination of neglected tropical diseases (4). A key pillar of this roadmap is the urgent need for the discovery and development of novel therapeutic agents targeting schistosomiasis. In this context, the identification of new chemical scaffolds with selective anthelmintic activity is a critical step toward diversifying the pipeline and reducing reliance on a single therapeutic option (5, 6).
Drug repurposing offers a cost-effective strategy to combat neglected tropical diseases, utilizing existing drugs with known safety profiles to expedite clinical use. Another innovative strategy is hybrid drug discovery, which merges pharmacophores from different bioactive molecules (7, 8). The goal is to enhance antischistosomal efficacy, reduce resistance, and enable multi-stage parasite targeting. By integrating distinct bioactive fragments into a single scaffold, these hybrids seek to overcome praziquantel’s limitations.
Herein, we present a hybrid design strategy that combines pharmacophoric elements from praziquantel, mirasan, and mefloquine to generate novel scaffolds with potential antischistosomal activity (Fig. 1). The rationale for compound selection was based on hybridization of pharmacophoric fragments derived from these molecules, representing distinct chemotypes previously associated with antiparasitic activity (9, 10). Praziquantel provides a rigid hydrophobic core and cyclic amide motif that interacts with calcium channels, whereas mirasan serves as a structural precursor for the development of hycanthone and oxamniquine, two classical DNA-intercalating antischistosomal agents (11, 12). Mefloquine contributes a quinoline-based lipophilic scaffold that perturbs parasite membrane stability (13). The imidazolidine-2,4-dione and isoindole-1,3-dione frameworks were chosen as versatile linkers to integrate these pharmacophores, enabling modulation of electronic and steric properties within a chemically accessible platform. All final compounds (1–7 and 9–12), except for compound 8, have been previously reported (9, 10, 14–17). The detailed description of the synthesis of compounds 1–12 is presented in the supplemental material (available at http://dx.doi.org/10.60731/ujcm.18122501).
Hybrid scaffold design strategy and chemical structures of the compounds evaluated in this study. (Top) Schematic representation of the pharmacophore-based design combining structural fragments from praziquantel, mefloquine, and mirasan to generate imidazolidine-2,4-dione derivatives (compounds 8, 9, 11, and 12). (Bottom) Chemical structures of all synthesized compounds (1–12), including isoindole-1,3-dione derivatives (1–5) and imidazolidine-2,4-dione derivatives (6–12). Compound 8 is novel; all other compounds correspond to previously reported structures. Full synthetic details and analytical data are provided in the supplemental material.
Adult Schistosoma mansoni worms (BH strain) were obtained from infected Swiss mice 49 days post-infection (18).
Each experiment was performed in triplicate using four pairs of adult worms per well, incubated in 24-well plates with 2 mL of RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C in a 5% CO_2_ atmosphere. In the initial screening, all compounds were tested at 50 µM for 72 h, and worm viability was assessed at 0, 24, 48, and 72 h under an inverted microscope. Parasite death was defined as the complete absence of movement for at least 1 minute following gentle mechanical stimulation with fine forceps (19). Compounds that reduced worm viability in this primary screen were subsequently tested across concentrations ranging from 6.25 to 50 µM to determine the 50% effective concentration (EC_50_) by non-linear regression of concentration–response data using GraphPad Prism 10 (20, 21). For cytotoxicity assays, Vero cells (1 × 10⁴ cells/well) were incubated with each compound for 24 h prior to MTT addition, and absorbance was measured at 570 nm (22). All experiments were performed in triplicate, and results are expressed as mean ± SD from three independent replicates.
Representative dose–response curves for the active compounds are provided in Fig. S1 (http://dx.doi.org/10.60731/ujcm.18122501). All active molecules induced progressive, concentration-dependent reductions in worm motility and viability without immediate paralysis, suggesting a slower onset of action distinct from the rapid spastic contraction produced by praziquantel. These observations support the hypothesis that the imidazolidine-2,4-dione scaffold acts through a mechanism different from that of praziquantel.
All compounds from the isoindole-1,3-dione series (1–5) were inactive at the tested concentration, despite variations in the substituents on the isoindole-1,3-dione ring (e.g., OCH_3_ or H) and differences in the length of the linker connecting the isoindole-1,3-dione to the amine moiety (containing pyridine, benzimidazole, or 1,2,3,4-tetrahydroisoquinoline groups). Among the imidazolidine-2,4-dione derivatives (6–12), compounds 6, 7, and 10 were also inactive. In contrast, compounds 8, 9, 11, and 12 induced marked reductions in worm viability (Table 1).
It is worth mentioning that 8, 9, and 11 have an imidazolidine-2,4-dione scaffold with a α-tetralinyl or 4-phenylcyclohexyl moiety, connected via an ethyl or propyl linker to a 3-trifluoromethylphenylpiperazine moiety. This structure–activity relationship analysis revealed that replacing the α-tetralin scaffold with β-tetralin (6 vs 9) or replacing the 3-trifluoromethyl group with a 3-fluoro group in the phenylpiperazine moiety (10 vs 11) in their structural counterparts resulted in the loss of biological activity against S. mansoni. Moreover, the substitution of the 2,3-dimethoxybenzylidene fragment into the imidazolidine-2,4-dione ring, along with the replacement of the 3-trifluoromethyl group with a 2,3-dichloro group (12), allowed for the retention of antischistosomal activity. Interestingly, the physicochemical properties of pharmacologically active hybrid compounds were similar (lipophilicity and water solubility), and all compounds agreed with the drug-likeness and Lipinski rule (see Table S1 at http://dx.doi.org/10.60731/ujcm.18122501).
These active compounds (8, 9, 11, and 12) were subsequently tested in concentration–response experiments. None of the compounds tested showed cytotoxicity in Vero cells at concentrations up to 200 µM. Consequently, selectivity indices for 8, 9, 11, and 12 were all above 10, indicating a favorable selectivity profile. Detailed results for all tested compounds, including EC_50_, 50% cytotoxic concentration against Vero cells, and SI values, are presented in Table 1. Although EC_50_ values in the 10–20 µM range are moderate, they are consistent with reported activity thresholds for early-stage antischistosomal hits (23, 24).
Taken together, these findings support the imidazolidine-2,4-dione scaffold as a promising platform for the development of new antischistosomal agents. The inactivity of the isoindole-1,3-dione series underlines the importance of scaffold selection in the early stages of drug discovery. Compounds 9 and 12, in particular, demonstrated high potency and selectivity, making them strong candidates for further pharmacological and mechanistic studies.
Further optimization of these hybrid derivatives, including an expanded structure–activity relationship analysis of compound 8 and its analogs, is currently underway in our laboratory. These studies aim to enhance potency while preserving the favorable selectivity and physicochemical characteristics demonstrated in the present work.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Buonfrate D, Ferrari TCA, Akim Adegnika A, Russell Stothard J, Gobbi FG. 2025. Human schistosomiasis. Lancet 405:658–670. doi:10.1016/S 0140-6736(25)00768-839986748 · doi ↗ · pubmed ↗
- 2Morais CS, Mengarda AC, Miguel FB, Enes KB, Rodrigues VC, Espírito-Santo MCC, Siyadatpanah A, Wilairatana P, Couri MRC, de Moraes J. 2021. Pyrazoline derivatives as promising novel antischistosomal agents. Sci Rep 11:23437. doi:10.1038/s 41598-021-02792-034873205 PMC 8648852 · doi ↗ · pubmed ↗
- 3Mengarda Ana C, Iles B, F Longo JP, de Moraes J. 2022. Recent trends in praziquantel nanoformulations for helminthiasis treatment. Expert Opin Drug Deliv 19:383–393. doi:10.1080/17425247.2022.205147735264036 · doi ↗ · pubmed ↗
- 4World Health Organization. 2020. Ending the neglect to attain the sustainable development goals: a road map for neglected tropical diseases 2021–2030. WHO.
- 5Mengarda AC, Silva MP, Cirino ME, Morais TR, Conserva GAA, Lago JHG, Moraes J. 2021. Licarin A, a neolignan isolated from Nectandra oppositifolia Nees & Martt. (Lauraceae), exhibited moderate preclinical efficacy against Schistosoma mansoni infection. Phytother Res 35:5154–5162. doi:10.1002/ptr.718434089558 · doi ↗ · pubmed ↗
- 6Longo JPF, de Moraes J. 2025. Recent advances in nanoformulations for helminthiasis treatment. Expert Opin Drug Deliv 22:1861–1876. doi:10.1080/17425247.2025.256542640974615 · doi ↗ · pubmed ↗
- 7Rugel A, Tarpley RS, Lopez A, Menard T, Guzman MA, Taylor AB, Cao X, Kovalskyy D, Chevalier FD, Anderson TJC, Hart PJ, Lo Verde PT, Mc Hardy SF. 2018. Design, synthesis, and characterization of novel small molecules as broad range antischistosomal agents. ACS Med Chem Lett 9:967–973. doi:10.1021/acsmedchemlett.8b 0025730344901 PMC 6187409 · doi ↗ · pubmed ↗
- 8Dziwornu GA, Attram HD, Gachuhi S, Chibale K. 2020. Chemotherapy for human schistosomiasis: how far have we come? What’s new? Where do we go from here? RSC Med Chem 11:455–490. doi:10.1039/d 0md 00062 k 33479649 PMC 7593896 · doi ↗ · pubmed ↗