Calabanones A‒H, chromanone derivatives from the stem bark of Calophyllum calaba and their cytotoxic activities against cancer cells
Sutin Kaennakam, Fadjar Mulya, Manussada Ratanasak, Kitiya Rassamee, Pongpun Siripong, Yasuteru Shigeta, Preecha Phuwapraisirisan, Edwin R. Sukandar

TL;DR
Researchers discovered eight new chromanone compounds from a tropical plant and found some to be moderately effective against cancer cells.
Contribution
The discovery of eight new chromanone derivatives and the first report of chromanone–steroid hybrids in plants.
Findings
Compounds 7 and 8 are the first chromanone–steroid hybrids reported in plants.
Compounds 6 and 9 showed moderate cytotoxic activity against KB and HeLa S3 cancer cells.
Compounds 3–5 selectively inhibited KB cell growth with IC50 values between 18.77 and 27.06 μM.
Abstract
The genus Calophyllum (Calophyllaceae), distributed mainly in tropical regions, is rich in chromanone derivatives with diverse molecular structures that exhibit potential antimicrobial and anticancer effects. Phytochemical investigation of the stem bark of C. calaba collected in Thailand led to the discovery of eight previously undescribed chromanones, calabanones A–H (1‒8), and two known analogs, (‒)-isocalomembranone P (9) and (‒)-calomembranone P (10). The chemical structures of undescribed compounds were elucidated using spectroscopic analyses, particularly NMR and HRESIMS, while their absolute configurations were determined through ECD and NMR calculations combined with DP4+ probability analysis. Compounds 7 and 8 were identified as chromanone–steroid hybrids linked via an ester bond, representing the first report of such structures in plants. Cytotoxic evaluation of the isolated…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
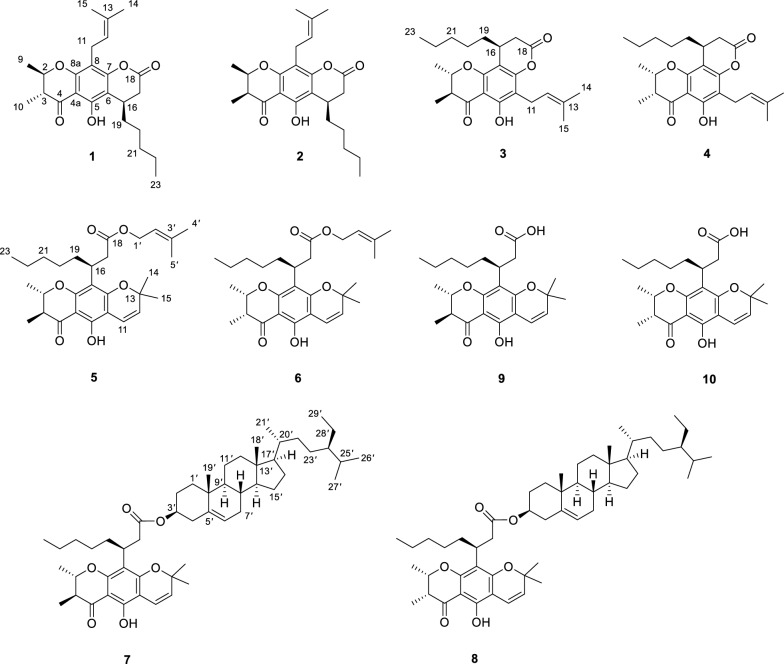 Figure 1
Figure 1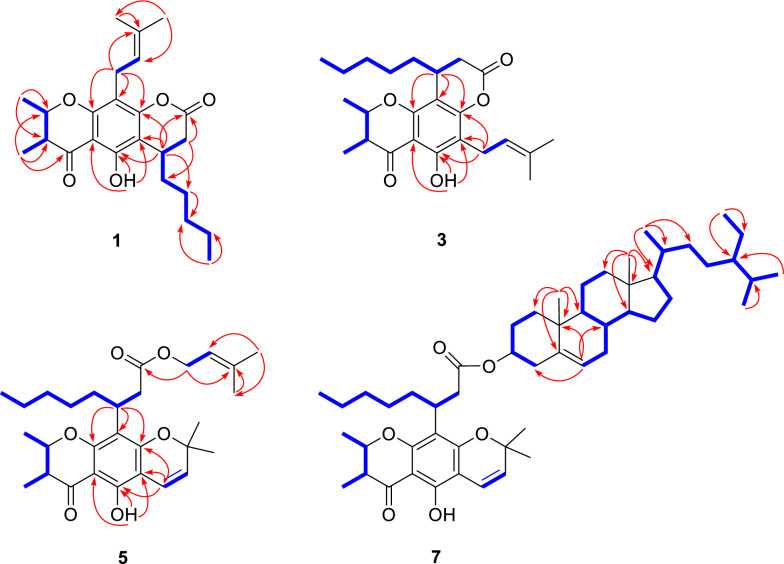 Figure 2
Figure 2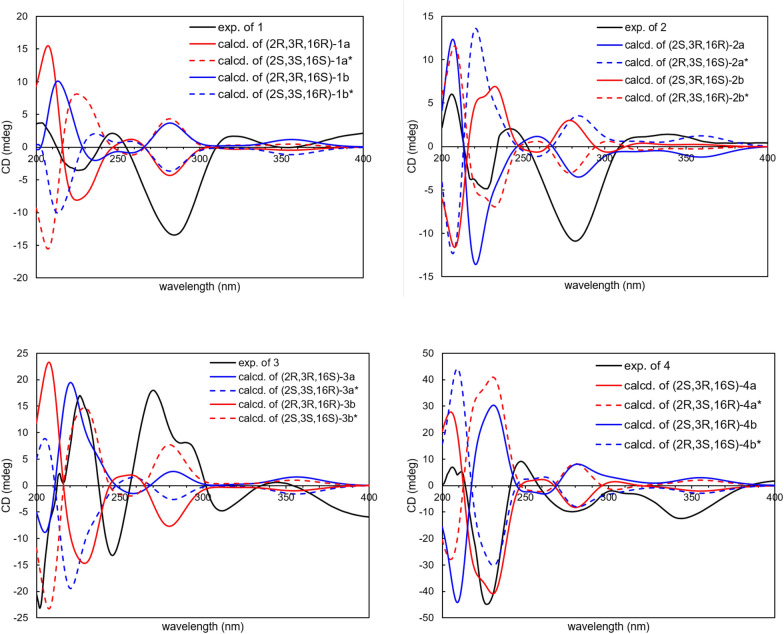 Figure 3
Figure 3 Figure 4
Figure 4- —http://dx.doi.org/10.13039/501100007345King Mongkut's University of Technology North Bangkok
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNatural Compound Pharmacology Studies · Phytochemistry and Biological Activities · Biological and pharmacological studies of plants
Introduction
Chromanones are among the major specialized metabolites of Calophyllum plants (Calophyllaceae), alongside xanthones and coumarins. They are biosynthesized via Claisen condensation of isovaleryl-CoA with three malonyl-CoA units, yielding a 2,3-dimethylchromanone scaffold [1]. The incorporation of isoprene units and either cinnamic acid or short-chain fatty acids (C_6_ or C_8_) onto the chromanone skeleton generates diverse chemical architectures through esterification and cyclization, resulting in pyranochromanones and pyranocoumarins [2]. Several chromanone derivatives have demonstrated promising pharmacological activities, including (+)-calanolide A and (+)-inophyllum B, both known for their antiviral potential. The former, originally isolated from C. lanigerum, strongly inhibits HIV-1 reverse transcriptase and has advanced to phase II clinical trials as an anti-HIV drug candidate, while the latter, the major constituent of C. inophyllum, is under preclinical evaluation, exhibiting a similar mechanism of action to calanolide A with inhibition at the nanomolar level [2, 3].
Calophyllum calaba L., locally known as “Thunghoon” in Thailand, grows in peat swamp and beach forests and is primarily distributed across Southeast Asia, Sri Lanka, and India [4]. Only limited phytochemical studies have been conducted on this species, including investigations of its stem bark, leaves, and roots from Sri Lanka, which revealed several bioactive metabolites such as chromanones, xanthones, biflavonoids, and triterpenoids [5–9]. In our previous work, cytotoxic prenylated xanthones were isolated from the roots of C. calaba collected in Thailand, with calaxanthone C exhibiting the strongest activity against five human cancer cell lines (KB, HeLa S3, HT-29, MCF-7, and HepG2), showing IC_50_ values below 5 μM [10]. Motivated by these results, we performed chemical screening using NMR analysis on extracts and fractions from other plant organs, which indicated that the stem bark extract contained characteristic proton and carbon signals of chromanone derivatives. Herein, we report the isolation, structure elucidation, and cytotoxic evaluation of chromanones obtained from the CH_2_Cl_2_ extract of C. calaba stem bark.
Results and discussion
Eight previously undercribed chromanones, calabanones A‒H (1–8), and two known analogs, (‒)-isocalomembranone P (9) and (‒)-calomembranone P (10) [1], were isolated from the CH_2_Cl_2_ extract of C. calaba stem bark through successive chromatographic separations, including silica gel, RP-C_18_, Sephadex LH-20, and radial chromatographic techniques. The structures of the previously undescribed specialized metabolites were elucidated by spectroscopic analyses (NMR and HRESIMS) and by comparison of their ^1^H and ^13^C NMR data with those of related compounds reported in the literature (Fig. 1).Fig. 1. Chromanones 1 − 10 isolated from the stem bark of C. calaba
Calabanone A (1) was obtained as pale yellow gum. Its molecular formula, C_24_H_32_O_5_, was established from the sodium adduct ion at m/z 423.2159 [M + Na]^+^ (calcd for C_24_H_32_O_5_Na^+^, 423.2142) in the HRESIMS, indicating nine degrees of unsaturation. The UV absorption maxima at 215, 284, and 348 nm suggested the presence of a chromanone chromophore, consistent with those reported for Calophyllum-derived chromanones [1, 11]. The ^1^H NMR spectrum (Table 1) showed resonances for a hydrogen-bonded hydroxy proton at δH 12.26 (OH-5) and two methyl doublets at δH 1.53 (3H, d, J = 6.4 Hz, H-9) and 1.22 (3H, d, J = 6.8 Hz, H-10). The ^1^H–^1^H COSY spectrum revealed correlations of a contiguous spin system of H-10/H-3 (δH 2.59, dq, J = 12.4, 6.8 Hz)/H-2 (δH 4.19, dq, J = 12.4, 6.4 Hz)/H-9. The HMBC correlations of H-9 and H-10 to C-2 (δC 79.1) and C-3 (δC 46.2), as well as H-10 to the carbonyl C-4 (δC 200.3), confirmed the presence of a 2,3-dimethylchromanone scaffold (Fig. 2). An octanoic acid ester attached to the aromatic ring was deduced from the ester carbonyl C-18 (δC 167.6), together with proton signals of a methine at δH 3.33 (1H, q, J = 6.8 Hz, H-16), a terminal methyl at δH 0.86 (3H, t, J = 6.8 Hz, H-23), and five sets of methylenes at δH 2.83 (1H, d, J = 15.6 Hz, H-17a) and 2.64 (1H, dd, J = 15.6, 6.8 Hz, H-17b), 1.54 and 1.44 (each 1H, overlap, H-19), 1.41 and 1.28 (each 1H, overlap, H-20), 1.26 (2H, overlap, H-21), and 1.28 (2H, overlap, H-22). The COSY cross-peaks of H-17/H-16/H-19/H-20/H-21/H-22/H-23 supported this assignment. The HMBC correlations of H-16 to C-18 and C-20 (δC 26.5), H-17 to C-19 (δC 34.2), H-20 to C-21 (δC 31.8), and H-23 to C-21 and C-22 (δC 22.6) further confirmed the side chain. Additionally, correlations of OH-5 with C-4a (δC 104.2), C-5 (δC 157.6), and C-6 (δC 107.5), and of H-16 with C-5, C-6, and C-7 (δC 156.3), indicated cyclization of the side chain to C-6 and C-7, forming a pyran-2-one ring. The 1D NMR data also indicated a prenyl unit, as evidenced by resonances of a methine at δH 5.16 (1H, t, J = 7.6 Hz, H-12)/ δC 121.9 (C-12), a methylene at δH 3.28 (2H, d, J = 7.6 Hz, H-11)/* δC 21.7 (C-11), two methyl singlets at δH 1.66 (3H, s, H-14)/ δC 25.9 (C-14) and δH 1.77 (3H, s, H-15)/ δC 17.9 (C-15), and a sp^2^ quaternary carbon at δC 132.1 (C-13). The HMBC correlations of H-11 with C-7, C-8 (δC 108.9), and C-8a (δC 158.4) established the prenyl group at C-8.Table 1^1^H (400 MHz) and ^13^C (100 MHz) NMR spectroscopic data of compounds 1‒4 in CDCl_3_No**12**34δH (J in Hz)δCδH (J in Hz)δCδH (J in Hz)δCδ*H (J in Hz)δ_C_24.19, dq (12.4, 6.4)79.14.57, qd (6.4, 3.2)76.54.19, dq (12.4, 6.4)79.34.57, qd (6.4, 3.2)76.632.59, dq (12.4, 6.8)46.22.60, qd (7.2, 3.2)44.72.60, dq (12.4, 6.8)46.22.60, qd (7.2, 3.2)44.74200.3202.1199.8201.74a104.2103.6104.1103.55157.6157.9159.9160.36107.5107.6110.4110.57156.3156.4156.0155.98108.9109.0105.8105.88a158.4158.2156.2156.391.53, d (6.4)19.81.41, d (6.4)16.51.51, d (6.4)19.71.40, d (6.4)16.4101.22, d (6.8)10.21.20, d (7.2)9.41.23, d (6.8)10.41.18, d (7.2)9.5113.28, d (7.6)21.73.28, d (7.6)21.73.32, d (7.2)21.43.32, d (7.2)21.4125.16, t (7.6)121.95.15, t (7.6)121.95.19, t (7.2)121.65.20, t (7.2)121.613132.1132.2132.5132.5141.66, s25.91.66, s25.91.66, s25.91.67, s25.9151.77, s17.91.77, s17.91.79, s18.01.79, s18.0163.33, q (6.8)28.73.30, q (6.8)28.73.27, q (6.8)28.83.28, qd (6.4, 2.0)28.8172.83, d (15.6)2.64, dd (15.6, 6.8)34.02.82, d (15.6)2.63, dd (15.6, 6.8)33.92.81, d (15.6)2.67, dd (15.6, 6.8)34.32.79, dd (15.6, 2.0)2.66, dd (15.6, 6.4)34.518167.6167.6167.6167.5191.54^a^1.44^a^34.21.55^a^1.44^a^34.11.48^a^36.61.46, q (6.4)34.8201.41^a^1.28^a^26.51.40^a^1.28^a^26.51.40^a^1.29^a^26.51.37^a^1.27^a^26.6211.26^a^31.81.26^a^31.81.27^a^31.61.26^a^31.6221.28^a^22.61.28^a^22.61.29^a^22.61.28^a^22.7230.86, t (6.8)14.10.86, t (6.8)14.10.87, t (6.8)14.10.87, t (6.8)14.1OH-512.26, s12.18, s12.26, s12.17, s^a^Overlapping signals^b^Deduced from HMBC experimentsFig. 2Key COSY and HMBC correlations of compounds 1, 3, 5, and 7
The large coupling constant between H-2 and H-3 (^3^J2,3 = 12.4 Hz) favored a trans-2,3-dimethyl configuration, with both protons was in an axial relationship (Table 1). The small ^3^J16,17 coupling (6.8 Hz) between H-16 and H-17 suggested that the n-amyl unit at C-16 was axially oriented in the half-chair conformation of the pyran-2-one ring. However, due to the spatial separation between H-16 and H-2/H-3, the relative configuration of C-16 could not be unambiguosly assigned. Thus, the NMR calculation coupled with DP4+ probability analysis were performed for two possible conformers (2R,3R,16R-1a and 2R,3R,16S-1b) using the GIAO method at the B3LYP/6-31++ G(d,p) level. DP4+ statistical analysis indicated isomer 1a (16R) as the predominant conformer, with probabilities of 99.91% (^1^H and ^13^C data) (Additional file 1: Table S1). The absolute configuration was confirmed by quantum ECD calculations for the two conformers 1a and 1b and their enantiomeric pairs 2S,3S,16S-1a* and 2S,3S,16R-1b*, with the predicted spectrum of 1a closely matching the experimental data (Fig. 3). Accordingly, the absolute configuration of compound 1 was assigned as 2R,3R,16R.Fig. 3. Experimental and calculated ECD spectra of 1‒4
Calabanone B (2) was assigned the molecular formula C_24_H_32_O_5_, based on the sodium adduct ion at m/z 423.2137 [M + Na]^+^ (calcd for C_24_H_32_O_5_Na^+^, 423.2142) in HRESIMS. Comparison of its ^1^H and ^13^C NMR data with those of 1 revealed that the structural differences were confined to the 2,3-dihydro-2,3-dimethyl-γ-pyran-4-one moiety. The small ^3^J value (3.2 Hz) between H-2 and H-3 preferred a cis-2,3-dimethyl configuration (Table 1). To determine the relative configuration at C-16, NMR calculations were performed for two possible diastereomers (2S,3R,16R-2a and 2S,3R,16S-2b). DP4+ statistical analysis of the calculated ^1^H and ^13^C NMR data supported 2b as the correct conformer, with an overall probability of 94.35% (Additional file 1: Table S3). To further corroborate the NMR/DP4+ results, quantum ECD calculations were carried out for 2a and 2b and their corresponding enantiomers (2R,3S,16S-2a* and 2R,3S,16R-2b*). The calculated ECD spectrum of model 2b* closely matched the experimental data (Fig. 3), thereby confirming the absolute configuration of compound 2 as 2R,3S,16R (Fig. 3).
Calabanone C (3) was obtained as a pale yellow gum. The ( +)-HRESIMS spectrum displayed a protonated molecule [M + H]^+^ at m/z 401.2323 (calcd for C_24_H_33_O_5_^+^, 401.2323). The ^1^H and ^13^C NMR spectra of 3 closely resembled those of 1, except for variations in the aromatic carbon resoncances, suggesting a difference in side chain arrangement. In the HMBC spectrum, H-11 (δH 3.32, 2H, d, J = 7.2 Hz) correlated with C-5 (δC 159.9), C-6 (δC 110.4), and C-7 (δC 156.0), while H-16 (δH 3.27, 1H, q, J = 6.8 Hz) showed cross-peaks with C-7, C-8 (δC 105.8), and C-8a (δC 156.2). These correlations indicated that the prenyl unit was located at C-6 and the 2-pyranone moiety was fused at C-7 and C-8 of the chromamone skeleton (Fig. 2). The large coupling constant (^3^* J* = 12.4 Hz) between H-2 and H-3 supported a trans-2,3-dimethyl configuration with the protons in an axial orientation. The relative configuration at C-16 was further resolved by NMR calculations, which identified conformer 2R,3R,16R-3b as the most possible candidate with a DP4+ probability of 99.46% in both ^1^H and ^13^C NMR data (Additional file 1: Table S5), compared with its diastereomer 2R,3R,16S-3a in DP4+ analysis. To confirm the absolte configuration, ECD calculations were performed for models 3a and 3b as well as their enantiomeric pairs (2S,3S,16R-3a* and 2S,3S,16S-3b*). The calculated ECD spectrum of the conformer 3b* matched the experimental data, thereby establishing the absolute configuration of 3 as 2S,3S,16S (Fig. 3).
The molecular formula of calabanone D (4), C_24_H_32_O_5_, was deduced from its protonated molecule at m/z 401.2319 [M + H]^+^ (calcd for C_24_H_33_O_5_^+^, 401.2323) in HRESIMS. Analysis of the 2D NMR data revealed key HMBC and COSY correlations similar to those of 3. However, the small coupling constant between H-2 and H-3 (^3^J = 3.2 Hz) indicated a cis-2,3-dimethyl configuration (Table 1). DP4+ statistical analysis supported conformer 2S,3R,16S-4a as the most probable structure, with an overall probability of 99.12% in all H and C data (Additional file 1: Table S7), compared with its diastereomer 2S,3R,16R-4b. The absolute configuration of 4 was further assessed by ECD calculations performed for 4a and 4b and their enantiomeric pairs 2R,3S,16R-4a* and 2R,3S,16S-4b*, respectively. The experimental ECD curve were similar with those calculated for 4a and 4b*, with 4a being slightly favored due to the presence of an additional minor positive CE at approximately 302 nm as well as similar CE amplitudes at ~ 227 and 206 nm that were also observed in the experimental spectrum. Taken together with the high DP4+ probability for 4a, these results established the absolute configuration of compound 4 as 2S,3R,16S (Fig. 3).
Calabanone E (5) was isolated as a pale yellow gum. Its molecular formula, C_29_H_40_O_6_, was determined from the sodium adduct ion at m/z 507.2739 [M + Na]^+^ (calcd for C_29_H_40_O_6_Na^+^, 507.2717) in the HRESIMS, indicating 10 degrees of unsaturation. The ^1^H and ^13^C NMR spectra (Table 2) showed resonances for two olefinic protons at δH 6.61 (1H, d, J = 10.0 Hz, H-11) and 5.46 (1H, d, J = 10.0 Hz, H-12), two gem-dimethyls at δH 1.44 (each 3H, s, H_3_-14 and H_3_-15), and an oxygenated sp^3^ carbon at δC 78.2 (C-13), supporting the presence of a pyran unit. HMBC correlations of H-11 to C-5 (δC 157.1), C-6 (δC 102.7), and C-7 (δC 160.3) placed the pyran ring at C-6 and C-7. The 1D NMR data closely resembled those of isocalomembranone P (9) from C. membranaceum, except for differences in the fatty acid side chain [1]. The more shielded ester carbonyl at δC 173.4 (C-18) in 5 suggested and ester bond formation. Additional resonances, including an olefinic proton at δH 5.20 (1H, t, J = 7.2 Hz, H-2′), diastereotopic oxymethylene protons at δH 4.49 and 4.43 (each 1H, dd, J = 12.4, 7.2 Hz, H_2_-1′), two methyl protons at δH 1.70 (3H, s, H_3_-4′) and 1.63 (3H, s, H_3_-5′), and a quaternary sp^2^ carbon at δC 138.5 (C-3′), indicated a prenyl unit linked to the ester carbonyl, as confirmed by the HMBC correlation of H_2_-1′ to C-18 (Fig. 2). The large coupling constant (J = 12.4 Hz) between H-2 (δH 4.12) and H-3 (δH 2.52) supported a trans-diaxial relationship, with both methyl groups H_3_-9 and H_3_-10 in equatorial orientations. The experimental ECD spectrum (Additional file 1: Fig. S49) and specific rotation ( \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} = ‒46.2) of 5 was identical to that of (‒)-isocalomembranone P (9) [1], confirming the absolute configuration as 2S,3S,16S.Table 2^1^H (400 MHz) and ^13^C (100 MHz) NMR spectroscopic data of compounds 5‒8 in CDCl_3_No**567**8δH (J in Hz)δCδH (J in Hz)δCδH (J in Hz)δCδH (J in Hz)δ_C_24.12, dq (12.4, 6.4)79.04.50, qd (6.4, 3.2)76.14.13, dq (12.4, 6.4)78.94.52, qd (6.4, 3.2)76.132.52, dq (12.4, 6.8)45.92.52, qd (7.2, 3.2)44.42.51, dq (12.4, 6.8)45.92.54, qd (7.2, 3.2)44.44199.5201.3199.5201.34a102.0101.3102.0101.35157.1157.4157.1157.46102.7102.7102.7102.77160.3^b^160.6^b^160.4^b^160.4^b^8109.5109.4109.6109.58a160.1^b^160.0^b^160.0^b^160.0^b^91.49, d (6.4)19.71.38, d (6.4)16.51.50, d (6.4)19.71.39, d (6.4)16.5101.20, d (6.8)10.61.15, d (7.2)9.41.20, d (6.8)45.91.16, d (7.2)9.6116.61, d (10.0)115.96.60, d (10.0)115.86.61, d (10.0)116.06.61, d (10.0)115.9125.46, d (10.0)125.75.46, d (10.0)125.75.46, d (10.0)125.75.46, d (10.0)125.71378.278.578.278.3141.44, s28.51.44, s28.61.45, s28.61.45, s28.7151.44, s28.51.44, s28.51.45, s28.41.45, s28.4163.69, m31.13.70, m31.23.69, m31.23.67, m31.3172.79, dd (14.8, 8.4)2.64, dd (14.8, 6.8)39.22.80, dd (14.8, 8.4)2.65, dd (14.8, 7.2)39.12.78, dd (14.8, 8.4)2.60, dd (14.8, 6.8)39.42.80, dd (14.8, 8.4)2.60, dd (14.8, 6.8)39.418173.4173.4172.8172.8191.80, m1.53^a^33.41.76, m1.53^a^33.41.80^a^1.53^a^33.51.80^a^1.51^a^33.5201.10‒1.17^a^27.61.10‒1.16^a^27.61.10‒1.16^a^27.61.10‒1.16^a^27.6211.25^a^1.18^a^32.01.25^a^1.18^a^31.91.24^a^1.19^a^32.01.25^a^1.19^a^31.9221.22^a^22.71.23^a^22.81.23^a^22.71.23^a^22.8230.83, t (6.8)14.20.83, t (6.8)14.20.83, t (7.2)14.20.83, t (7.2)14.2OH-512.47, s12.39, s12.48, s12.39, s1′4.49, dd (12.4, 7.2)4.43, dd (12.4, 7.2)61.14.49, dd (12.4, 7.6)4.42, dd (12.4, 7.6)61.11.78^a^1.05^a^37.11.78^a^1.05^a^37.12′5.20, t (7.2)119.15.20, t (7.6)119.01.67^a^1.38^a^27.81.65^a^1.38^a^27.73′138.5138.54.49, m73.64.49, m73.64′1.70, s25.81.69, s25.82.22, m38.32.20, m38.25′1.63, s18.11.62, s18.0140.0139.96′5.32, d (6.0)122.65.31, d (5.2)122.67′1.94^a^32.11.94^a^32.08′1.41^a^32.01.41^a^32.09′0.91^a^50.20.90^a^50.210′36.736.711′1.46^a^1.41^a^21.21.45^a^1.41^a^21.212′1.98, m1.13^a^39.91.98, m1.13^a^39.913′42.542.514′0.98^a^56.90.98^a^56.915′1.56^a^1.04^a^24.41.56^a^1.03^a^24.416′1.83^a^1.25^a^28.51.82^a^1.25^a^28.417′1.09^a^56.21.09^a^56.218′0.67, s12.00.66, s12.019′0.97, s19.40.97, s19.420′1.33^a^36.31.33^a^36.321′0.91, d (6.4)19.20.91, d (6.4)19.222′1.31^a^1.00^a^34.11.31^a^1.00^a^34.123′1.14^a^26.31.14^a^26.324′0.90^a^46.00.91^a^46.025′1.65, m29.41.65, m29.426′0.83, d (7.2)20.00.83, d (7.2)20.027′0.81, d (7.2)18.90.81, d (7.2)18.928′1.26^a^23.31.25^a^23.329′0.83, t (7.2)12.10.83, t (7.2)12.1^a^Overlapping signals^b^Deduced from HMBC experiments
Calabanone F (6) possessed the molecular formula C_29_H_40_O_6_, as indicated by the sodium adduct ion at m/z 507.2736 [M + Na]^+^ (calcd for C_29_H_40_O_6_Na^+^, 507.2717) in the (+)-HRESIMS. Its 1D NMR spectra differed from those of 5 mainly in the orientation of the methyl group at C-2. The small coupling constant (^3^J2,3 = 3.2 Hz) indicated a cis-2,3-dimethyl configuration (Table 2). The experimental ECD spectrum of 6 (Additional file 1: Fig. S49) closely matched that of (‒)-calomembranone P (10) [1], supported by their similar negative specific rotatations, thereby establishing the absolute configuration of 6 as 2S,3R,16S.
The molecular formula of calabanone G (7) was determined as C_53_H_80_O_6_ from the protonated molecule at m/z 813.6028 in ( +)-HRESIMS (calcd for C_53_H_81_O_6_^+^, 813.6028). The ^1^H and ^13^C spectra indicated the same chromanone skeleton as in 5 but with a different ester moiety (Table 2). The absence of prenyl resonances, present in 5, was replaced by characteristic signals of β-sitosterol [12]. 2D NMR data confirmed the steroid unit, including its typical contiguous ^1^H‒^1^H spin system and HMBC correlations, such as olefinic proton H-6′ (δH 5.32, d, J = 6.0 Hz) to C-4′ (δC 38.3), C-8′ (δC 32.0), and C-10′ (δC 36.7) and six methyl resonances (H_3_-19′, H_3_-18′, H_3_-21′, H_3_-26′, H_3_-27′, and H_3_-29′) to the expected carbons of the steroid skeleton (Fig. 2). Although no direct HMBC correlation confirming the ester linkage between the 3′-OH of β-sitosterol and the chromanone carbonyl C-18, the downfield signals at δC 73.6 (C-3′) (ΔδC + 1.8 ppm) and the shielded carbonyl at δC 172.8 (C-18) (ΔδC ‒6.6 ppm), together with the HRESIMS data, supported a chromanone‒steroid hybrid linked via an ester bond [1, 12]. The ECD spectrum of 7 (Additional file 1: Fig. S49) closely matched that of (‒)-isocalomembranone P (9) [1], establishing the absolute configuration of 7 as 2S,3S,16S.
HRESIMS analysis of calabanone H (8) showed a sodium adduct ion at m/z 835.5810 [M + Na]^+^ (calcd for C_53_H_80_O_6_Na^+^, 835.5847), consistent with a chromanone‒steroid hybrid analogous to 7. 1D NMR data confirmed that 7 and 8 were diastereomers, differing only in the 2,3-dimethyl configuration. The small coupling constant between H-2 and H-3 (^3^* J* = 3.2 Hz), supported a cis-relationship of the two protons (Table 2). Comparison of the experimental ECD spectrum of 8 (Additional file 1: Fig. S49) with that of (‒)-calomembranone P (10) [1] confirmed the absolute configuration of 8 as 2S,3R,16S.
Calophyllum chromanones are predominantly characterized by substitution with side chains such as C_8_-fatty acids, as observed in compounds 9 and 10, as well as cinnamic acid, as in calomembranones L–O isolated from C. membranaceum [1]. Intramolecular cyclization between the carboxylic acid group and a hydroxy group on the aromatic ring can further generate coumarin-like structures, as shown in compounds 1–4, which represent another major class of derivatives within this genus [2]. In addition to cyclization, esterification of the carboxylic acid moiety is also a frequent structural modification among Calophyllum metabolites [2]. Previously reported esterifications were largely limited to simple methyl chromanone esters, such as calomembranones E–G [2, 11]. The identification of chromanones bearing prenyl esters (5 and 6) and chromanone–steroid hybrids linked through an ester bond (7 and 8) therefore represents, to the best of our knowledge, the first report of such structural motifs in plants. In general, naturally occurring triterpenoid or stereoid hybrids connected to other metabolites via ester linkage are rare. Representative examples include myrtphlotritins, phloroglucinol–triterpenoid adducts isolated from Leptospermum scoparium and Xanthostemon chrysanthus (Myrtaceae) [13] and the triterpenoid–chromone hybrids cimitriteromones originally identified from the rhizomes of Actaea cimicifuga (Ranunculaceae) [14]. Ester functionalities in natural products can sometimes arise as artifacts, particularly when carboxylic acid–containing compounds undergo esterification in the presence of alcohols, such as the formation of methyl or ethyl esters from methanol or ethanol solvents during isolation [15]. As the crude plant extract in this study was entirely used during the isolation process and thus unavailable for direct verification, acid-catalyzed esterification reactions were carried out using β-sitosterol and (–)-calomembranone P (10) to assess whether compound 8 could be an artifact. These reaction mixtures were refluxed in methanol for 24 h using either acetic acid or H_2_SO_4_ as catalysts. TLC analysis revealed no formation of compound 8 under either condition, suggesting that the chromanone–steroid hybrids are unlikely to be artifacts of the isolation process (Additional file 1: Fig. S51).
Previous studies have shown that chromanones isolated from Calophyllum species exhibited potent cytotoxic activities against various cancer cell lines, including KB and HeLa cells. Notable examples included caloteysmannic acid, isocalolongic acid, and apetalic acid derivatives, which displayed IC_50_ values below 10 μM [16, 17]. Based on these reports, the cytotoxic activities of all isolated compounds in this work were evaluated using an MTT assay against KB and HeLa S3 cells, together with the HepG2 cancer cell line, and the results are summarized in Table 3. Compounds 6 and 9 exhibited moderate activity against KB and HeLa S3 cells, with IC_50_ values ranging from 12.71 to 25.50 μM. Compounds 3‒5 selectively inhibited the growth of KB cells, showing IC_50_ values between 18.77 and 27.06 μM. The remaining compounds displayed weak cytotoxic effects, whereas those containing a sitosterol moiety (7 and 8) were inactive, with IC_50_ values exceeding 100 μM. Notably, chromanones apetalic acid, isoapetalic acid, and their methyl esters isolated from C. blancoi exhibited stronger cytotoxic activities against KB and HeLa cells [17] despite their close structural similarity to compounds 5, 6, 9, and 10. The key structural difference lies in the length of the fatty acid side chain, with the former bearing C_6_-fatty acids, whereas compounds 5, 6, 9, and 10 contain C_8_-fatty acids. A similar trend was observed for the derivatives with cyclized fatty acid. Cyclization of the fatty acid moiety in apetalic acid and isoapetalic acid retained potent activity (IC_50_ < 10 μM) [17], in contrast to their analogs 3 and 4. These preliminary structure–activity relationship (SAR) observations suggest that shorter fatty acid side chains play a crucial role in enhancing cytotoxic activity, possibly due to increased hydrophilicity. This interpretation is further supported by the lack of cytotoxicity observed for compounds 7 and 8, which bear bulky and nonpolar steroidal moieties. Nevertheless, additional chromanones with related functional groups are required for cytotoxic screening to strengthen this preliminary SAR analysis, and further evaluations against a broader panel of cancer cell lines as well as noncancerous cells are necessary to assess both potency and selectivity.Table 3. Cytotoxic activities^a^ of the isolated compounds after 72 h of treatmentCompoundIC_50_ ± SD *(*µM)KBHela S3HepG2141.50 ± 1.5356.47 ± 5.4041.40 ± 4.59258.13 ± 7.1084.52 ± 2.08 > 100321.64 ± 3.7251.11 ± 7.7488.50 ± 7.02427.06 ± 3.1076.72 ± 7.8284.15 ± 1.04518.77 ± 0.6730.24 ± 3.1491.79 ± 2.49612.71 ± 0.6016.63 ± 2.0252.01 ± 2.147 > 100 > 100 > 1008 > 100 > 100 > 100917.95 ± 0.5025.50 ± 2.0564.47 ± 2.621032.08 ± 2.9958.32 ± 3.8280.25 ± 9.19Doxorubicin^b^0.02 ± 0.000.14 ± 0.021.07 ± 0.16^a^Results are expressed as the mean values of three independent experiments ± SD. KB: human epidermoid carcinoma; Hela S3: human cervical carcinoma; HepG2: human hepatocellular carcinoma^b^Doxorubicin was used as the positive control. Note: IC_50_ ≤ 10 μM = good cytotoxicity; 10 μM < IC_50_ ≤ 30 μM = moderate cytotoxicity; 30 μM < IC_50_ ≤ 100 μM = weak cytotoxicity. The other compounds were inactive (IC_50_ > 100 µM)
Conclusions
In summary, eight previously undescribed chromanones, calabanones A–H (1–8), along with the known compounds (‒)-isocalomembranone P (9) and (‒)-calomembranone P (10), were isolated from the CH_2_Cl_2_ extract of the stem bark of C. calaba collected in Thailand. Notably, calabanones G and H represent the first naturally occurring chromanone–steroid hybrids, thereby expanding the chemical diversity and potential chemotaxonomic markers of Calophyllum species. In addition, calabanone F and (‒)-isocalomembranone P demonstrated moderate cytotoxicity against KB and HeLa S3 cell lines, with IC_50_ values below 30 μM, whereas the remaining compounds displayed weak or no activity against the tested cancer cell lines.
Experimental
General experimental procedures
Optical rotations were determined using a JASCO P-1010 polarimeter (JASCO Corporation, Easton, MD, USA). ECD spectra were obtained with a JASCO J-815 spectropolarimeter (JASCO Corporation). NMR spectra were measured on Bruker AVANCE 400 spectrometer (Bruker, Rheinstetten, Germany) using CDCl₃ as solvent (Merck, Darmstadt, Germany). HRMS data acquisition was performed on a Bruker MICROTOF model mass spectrometer (Bruker) and an Agilent 1260 series HPLC system coupled to a 6540 UHD accurate-mass QTOF mass spectrometer (Agilent Technologies, Waldbronn, Germany). Column chromatography was carried out using silica gel (70–230 mesh, Merck) and Sephadex LH-20 (25–100 μm, GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Radial chromatography was performed on a Chromatotron model 7924 T (Harrison Research, Palo Alto, CA, USA) equipped with silica gel 60 GF_2_₅₄ containing gypsum (≤ 55 μm, Merck). TLC analysis was conducted on silica gel 60 F_2_₅₄ plates (0.2 mm, Merck). Spots were initially observed under UV light at 254 and 366 nm, followed by staining with p-anisaldehyde reagent and subsequent heating to enhance visualization.
Plant material
The stem bark of C. calaba was collected in April 2023 from Buachet District, Surin Province, Thailand (14°33′24″ N, 104°00′56″ E). The species was identified by Dr. Suttira Sedlak, and a voucher specimen (Khumkratok no. 1–13) was deposited at the Walai Rukhavej Botanical Research Institute, Mahasarakham University.
Extraction and isolation
The air-dried stem bark of C. calaba (5.0 kg) was extracted by maceration with CH_2_Cl_2_ (3 × 10 L) at room temperature for three consecutive 3-day periods. Removal of the solvent yielded a crude extract (125.0 g), which was suspended in MeOH and partitioned with hexanes to obtain a hexanes-soluble fraction (64.0 g). This fraction was subjected to silica gel column chromatography using a gradient of hexanes–CH_2_Cl_2_ (100:0 to 0:100), affording eight fractions (A–H). Fraction A (8.6 g) was further chromatographed on silica gel with hexanes–CH_2_Cl_2_ (100:0 to 0:100) to yield subfractions A1–A7. Subfraction A2 (511.2 mg) was separated on a Sephadex LH-20 column with CH_2_Cl_2_–MeOH (50:50) to provide A2.1–A2.3. Purification of subfraction A2.3 (103.0 mg) by Chromatotron with hexanes–acetone (95:5) afforded compounds 3 (9.5 mg) and 4 (10.2 mg). Subfraction A3 (3.8 g) was fractionated on Sephadex LH-20 with CH_2_Cl_2_–MeOH (50:50) to give A3.1–A3.5. Further separation of A3.2 (712.0 mg) on a silica gel column using an isocratic hexanes–CH_2_Cl_2_ (50:50) system produced A3.2.1–A3.2.6. Compounds 1 (12.6 mg) and 2 (13.2 mg) were purified from subfractions A3.2.1 (108.0 mg) and A3.2.2 (113.5 mg), respectively, using Chromatotron with hexanes–EtOAc (95:5). Subfraction A3.2.6 (201.6 mg) was also purified under the same conditions with hexanes–acetone (95:5), yielding compounds 5 (4.8 mg), 6 (4.3 mg), 9 (3.8 mg), and 10 (3.2 mg). Fraction B (4.4 g) was separated on silica gel column chromatography with a gradient of n-hexane–CH_2_Cl_2_ (100:0 to 0:100), affording B1–B3. Subfractions B3 (821.0 mg) was subjected to Sephadex LH-20 using CH_2_Cl_2_–MeOH (50:50), yieldeing B3.1–B3.3. Purification of subfraction B3.1 (150.3 mg) by Chromatotron with hexanes–EtOAc (90:10) afforded compounds 7 (6.3 mg) and 8 (7.2 mg).
Calabanone A (1): Pale yellow gum; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} ‒40.0 (c 0.10, CHCl_3_); UV (MeOH) λ_max_ (log ε) 215 (3.3), 284 (1.7), 348 (0.9) nm; ECD (c 0.001, MeOH) λ_max_ (Δɛ) 204 (+ 3.7), 225 (‒3.5), 246 (+ 2.1), 284 (‒13.4), 321 (+ 1.7) nm, 354 (‒0.1); ^1^H (400 MHz, CDCl_3_) and ^13^C NMR (100 MHz, CDCl_3_) spectroscopic data, see Table 1; and HRESIMS m/z 423.2159 [M + Na]^+^ (calcd for C_24_H_32_O_5_Na^+^, 423.2142).
Isocalabanone A (2): Pale yellow gum; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} ‒25.0 (c 0.10, CHCl_3_); UV (MeOH) λ_max_ (log ε) 215 (2.6), 285 (1.3), 350 (0.8) nm; ECD (c 0.001, MeOH) λ_max_ (Δɛ) 206 (+ 6.1), 226 (‒4.9), 242 (+ 3.8), 285 (‒10.6) nm; ^1^H (400 MHz, CDCl_3_) and ^13^C NMR (100 MHz, CDCl_3_) spectroscopic data, see Table 1; and HRESIMS m/z 423.2137 [M + Na]^+^ (calcd for C_24_H_32_O_5_Na^+^, 423.2142).
Calabanone B (3): Pale yellow gum; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} ‒72.7 (c 0.10, CHCl_3_); UV (MeOH) λ_max_ (log ε) 219 (4.5), 279 (2.6), 349 (1.2) nm; ECD (c 0.001, MeOH) λ_max_ (Δɛ) 202 (‒23.1), 226 (+ 17.0), 246 (‒13.2), 270 (+ 17.8), 311 (‒4.8), 345 (+ 0.6) nm; ^1^H (400 MHz, CDCl_3_) and ^13^C NMR (100 MHz, CDCl_3_) spectroscopic data, see Table 1; and HRESIMS m/z 401.2323 [M + H]^+^ (calcd for C_24_H_33_O_5_^+^, 401.2323).
Isocalabanone B (4): Pale yellow gum; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} ‒14.5 (c 0.10, CHCl_3_); UV (MeOH) λ_max_ (log ε) 217 (4.5), 282 (2.5), 350 (1.1) nm; ECD (c 0.001, MeOH) λ_max_ (Δɛ) 206 (+ 7.0), 227 (‒44.8), 247 (+ 9.2), 278 (‒9.8), 302 (‒1.9), 344 (‒12.4) nm; ^1^H (400 MHz, CDCl_3_) and ^13^C NMR (100 MHz, CDCl_3_) spectroscopic data, see Table 1; and HRESIMS m/z 401.2319 [M + H]^+^ (calcd for C_24_H_33_O_5_^+^, 401.2323).
Calabanone C (5): Pale yellow gum; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} ‒46.2 (c 0.10, CHCl_3_); UV (MeOH) λ_max_ (log ε) 225 (2.5), 264 (2.9), 297 (1.8), 353 (0.9) nm; ECD (c 0.001, MeOH) λ_max_ (Δɛ) 210 (‒4.6), 227 (+ 9.5), 268 (‒2.7), 308 (+ 6.9) nm; ^1^H (400 MHz, CDCl_3_) and ^13^C NMR (100 MHz, CDCl_3_) spectroscopic data, see Table 2; and HRESIMS m/z 507.2739 [M + Na]^+^ (calcd for C_29_H_40_O_6_Na^+^, 507.2717).
Isocalabanone C (6): Pale yellow gum; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} ‒24.5 (c 0.10, CHCl_3_); UV (MeOH) λ_max_ (log ε) 227 (3.9), 251 (3.2), 318 (2.0), 359 (1.2) nm; ECD (c 0.001, MeOH) λ_max_ (Δɛ) 205 (‒14.3), 214 (+ 4.0), 228 (‒7.9), 275 (+ 3.0), 303 (‒6.4) nm; ^1^H (400 MHz, CDCl_3_) and ^13^C NMR (100 MHz, CDCl_3_) spectroscopic data, see Table 2; and HRESIMS m/z 507.2736 [M + Na]^+^ (calcd for C_29_H_40_O_6_Na^+^, 507.2717).
Calabanone D (7): Pale yellow gum; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} ‒46.0 (c 0.10, CHCl_3_); UV (MeOH) λ_max_ (log ε) 274 (1.9), 297 (1.2), 353 (0.7) nm; ECD (c 0.001, MeOH) λ_max_ (Δɛ) 224 (‒6.0), 247 (‒3.4), 265 (‒5.6), 301 (+ 2.0) nm; ^1^H (400 MHz, CDCl_3_) and ^13^C NMR (100 MHz, CDCl_3_) spectroscopic data, see Table 2; and HRESIMS m/z 813.6028 [M + H]^+^ (calcd for C_53_H_81_O_6_^+^, 813.6028).
Isocalabanone D (8): Pale yellow gum; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$${[\alpha ]}_{D}^{20}$$\end{document} ‒47.4 (c 0.10, CHCl_3_); UV (MeOH) λ_max_ (log ε) 274 (2.1), 298 (1.2), 363 (0.8) nm; ECD (c 0.001, MeOH) λ_max_ (Δɛ) 224 (+ 6.8), 239 (‒0.3), 263 (+ 4.0), 295 (‒3.4), 317 (+ 2.9) nm; ^1^H (400 MHz, CDCl_3_) and ^13^C NMR (100 MHz, CDCl_3_) spectroscopic data, see Table 2; and HRESIMS m/z 835.5810 [M + Na]^+^ (calcd for C_53_H_80_O_6_Na^+^, 835.5847).
ECD and NMR chemical shift calculations
The 3D molecular structures of two diastereomers for each compound (1‒4), differing only in the configuration at C-16, were initially constructed using Chem3D 19.1 software. The diastereomeric pairs included 2R,3R,16R-1a and 2R,3R,16S-1b, 2S,3R,16R-2a and 2S,3R,16S-2b, 2R,3R,16S-3a and 2R,3R,16R-3b, and 2S,3R,16S-4a and 2S,3R,16R-4b). These structures were subsequently subjected to conformational analysis using MOE software with the MMFF94 force field and Monte Carlo search [18]. The lowest-energy conformers contributing to more than 1.0% of the Boltzmann population were optimized by DFT at the B3LYP/6-31++ G(d,p) level. NMR chemical shifts were calculated using the GIAO method at the same level of theory. Vibrational frequency analysis confirmed the absence of imaginary modes. Shielding tensors were evaluated by the DP4+ probability method with Boltzmann weighting [19]. Chemical shifts were further refined with a multi-standard approach, using methanol for sp^3^ carbons and benzene for sp^2^ carbons [20, 21]. The optimized conformers were also subjected to ECD calculations. ECD spectra were simulated using TD-DFT B3LYP/6-31++ G(d,p) incorporating polarizable continuum model (PCM) with methanol as solvent, considering 50 excited states. The spectra were visualized in SpecDis 1.71 (σ = 0.2) with final spectra obtained by Boltzmann-weighted averaging [22]. The theoretical ECD spectra of the related enantiomers (conformers 1a*, 1b*, 2a*, 2b*, 3a*, 3b*, 4a*, and 4b*) were generated by applying mirror-image inversion to the calculated spectra. All calculations were carried out with Gaussian16 program [23].
Cytotoxicity assay
All the isolated compounds (1‒10) were tested for cytotoxic activities against three human cancer cell lines, epidermoid carcinoma (KB), cervical carcinoma (HeLa S3), and hepatocellular carcinoma (HepG2), following a reported protocol using MTT assay [24]. Doxorubicin served as the positive control, while 0.1% DMSO was used for vehicle controls. IC_50_ values were derived from dose–response curves as the concentration reducing cell viability by 50% relative to untreated cells. Results are presented as mean ± S.D. from three independent experiments.
Supplementary Information
Additional file 1 (PDF 5435 KB)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Forest Botany Division, Department of National Parks, Wildlife and Plant Conservation. e-Flora of Thailand. 2025. https://botany.dnp.go.th/eflora/floraspecies.html?tdcode=07182. Accessed 28 September 2025.