Electrochemical Synthesis of 1,2-Substituted N‑Amido Benzimidazoles by Reduction of Nitroarenes
Daniel Doellerer, Aaron Schüll, Thomas Weyhermüller, Sebastian B. Beil, Siegfried R. Waldvogel

TL;DR
This paper introduces a new electrochemical method to efficiently synthesize a rare type of benzimidazole compound with potential medical uses.
Contribution
A novel electrochemical protocol for synthesizing 1,2-substituted N-amido benzimidazoles with high yields and functional group tolerance.
Findings
The electrochemical method achieves target compounds in up to 89% yield.
The protocol is safe, reliable, and tolerant to various functional groups.
Abstract
Despite the vast number of reports on benzimidazoles, 1,2-substituted N-amido benzimidazoles remain an underrepresented and scarcely accessible compound class with promising pharmacological relevance. We present a safe, reliable electrochemical protocol that provides easy access to those structures. The reaction exhibits broad functional group tolerance and affords the target compounds in ≤89% yields.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
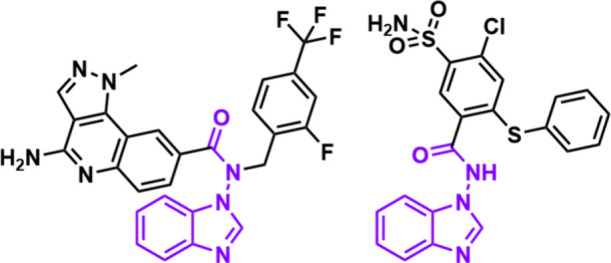 Figure 1
Figure 1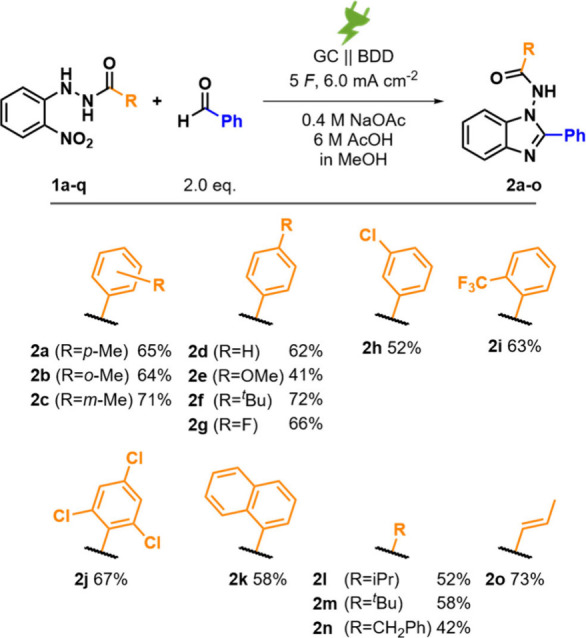 Figure 2
Figure 2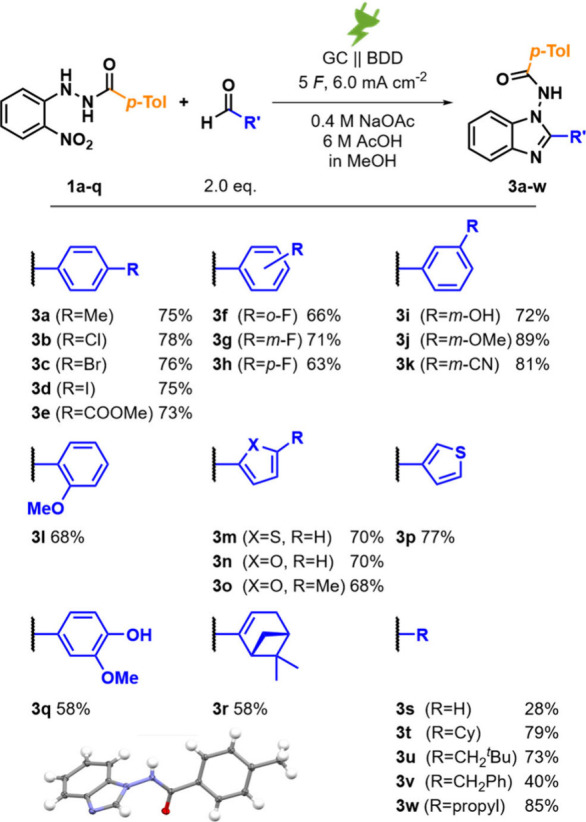 Figure 3
Figure 3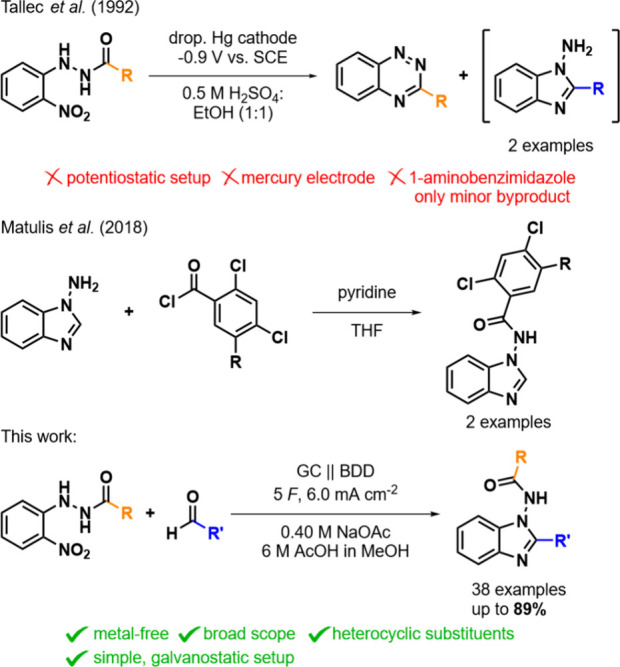 Figure 4
Figure 4 Figure 5
Figure 5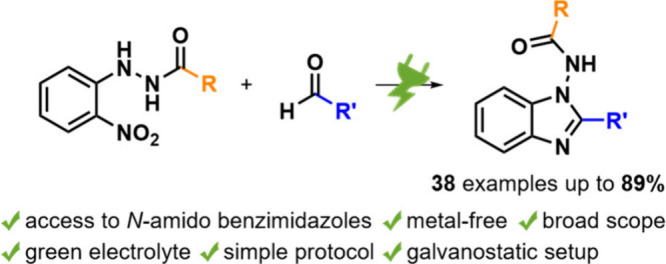 Figure 6
Figure 6 Figure 7
Figure 7- —Deutsche Forschungsgemeinschaft10.13039/501100001659
- —Fraunhofer-Gesellschaft10.13039/501100003185
- —Max-Planck-Gesellschaft10.13039/501100004189
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRadical Photochemical Reactions · Nanomaterials for catalytic reactions · Multicomponent Synthesis of Heterocycles
About 59% of small molecule drugs approved by the Food and Drug Administration (FDA) contain at least one nitrogen-based heterocycle, where newly approved derivatives involve as much as 82% N-heterocycles. ?,? Among these active pharmaceutical ingredients (APIs), benzimidazole moieties are common structural motifs that lead to a variety of therapeutic applications. ?−? ? ? Examples of drugs containing at least one benzimidazole are the well-known proton pump inhibitors such as omeprazole,? carbendazim, a commonly used fungicide,? second-generation antihistamine astemizole,? and telmisartan, an antihypertensive API. ?,? All FDA-approved benzimidazole-based drugs are substituted at position 2, and almost half of them (46%) are substituted in position 1, as well.? The conventional synthesis of benzimidazoles usually involves a condensation of o-phenylenediamines with different carbonyl components in a cyclo-condensation reaction. ?,? Alternative transformations usually start with o-nitroaniline. Metal catalysts and/or stoichiometric amounts of reducing agents are often used cogenerating large amounts of waste, which leads to bad atom economy.? Synthetic approaches toward N-amido benzimidazoles have only scratched the surface. Very few syntheses by direct reaction of N-amino benzimidazoles and acid anhydrides or acid chlorides have been reported, whereby the scope was limited to alkyl-substituted benzimidazoles (Scheme, middle). ?−? ? However, those few examples of N-amido benzimidazoles displayed high biological reactivity as protein arginine methyltransferase (PRMT) 5 or carbonic anhydrase VA inhibitors, which are usually upregulated in tumors or leukemia (see Figure). ?,? In contrast, aryl-substituted N-amido benzimidazoles have so far been reported only as minor byproducts during the synthesis of 1,2,4-benzotriazines (Scheme, top)? or when using the expensive and radioactive lanthanoid samarium.?
Electro-organic synthesis is a valuable alternative to traditional chemistry, circumventing those disadvantages. ?−? ? ? ? The use of electric current as a traceless oxidizing or reducing agent circumvents the formation of reagent waste. Furthermore, electrochemical reactions are often conducted under mild conditions, increasing functional group tolerance and saving energy by means of avoiding heating. ?,? Moreover, electro-organic transformations are considered to be inherently safe, since switching off the electrical current ceases the reaction almost immediately and thermal runaway reactions can be circumvented. ?,?
Our group established the electrochemical reduction of nitro groups as a promising and easy-to-use method for the synthesis of a large variety of N-heterocycles; ?,? among others, five-membered heterocycles like triazoles? and 2,1-benzisoxazoles ?,? have been synthesized. Since N-oxy and N-hydroxy heterocycles have shown interesting pharmacological properties and are often discussed as metabolites,? our group has reported a number of transformations with conditions, specifically chosen to yield these motifs. Among others, quinoline N-oxides,? 1-hydroxy quinazolinones,? 4-hydroxy-benzo[e]-1,2,4-thiadiazine-1,1-dioxides,? 1-hydroxyquin-ol-4-ones,? and cyclic hydroxamic acids? have been synthesized in this way.
Recently, the electro-organic synthesis of benzimidazoles has been realized by some groups in an entirely different way. In 2021, a method by Xu et al. was reported to synthesize benzimidazoles by electrochemical dehydrogenative cyclization of amidines.? Another approach for the electrochemical synthesis of benzimidazoles utilizing the oxidation of an alcohol for the generation of the carbonyl component is needed for the reaction while reducing a nitro group on the cathode. ?,? To the best of our knowledge, an electrochemical synthesis of N-amido benzimidazoles has not been reported. However, in 1992, Tallec et al. observed traces of 1-amino benzimidazoles as byproducts, while investigating the electrochemical synthesis of 1,2,4-benzotriazines from o-nitro phenyl hydrazides (Scheme, top).?
We present the first electrochemical synthesis of 2-substituted N-amido benzimidazoles from o-nitrophenyl hydrazides in a simple-to-operate and metal-free galvanostatic setup. The method tolerates a broad variety of functional groups such as double bonds, halo substituents, nitriles, alcohols, and other heterocycles, giving access to the desired benzimidazoles substituted in position 2 (Scheme bottom). To optimize the electrolysis conditions, o-nitro phenylhydrazide 1a was chosen as a test substrate to react with benzaldehyde to form benzimidazole 2a. 1a was synthesized in a single step from 2-nitrophenylhydrazine and 4-methylbenzoyl chloride (see the Supporting Information for experimental details).? To avoid the oxidation of the used aldehyde in the reaction, a divided cell setup was chosen for the electrochemical reduction of the nitro group. An applied charge slightly above the theoretical value of 4 F proved to be beneficial for the reaction, leading to the optimized conditions of 5 F with 6 mA cm^–2^ using a glassy carbon (GC) anode and boron-doped diamond (BDD) cathode, ?,? reacting 2 equiv of aldehyde with the respective hydrazide in a sodium acetate (0.4 M) and acetic acid (6 M) in a MeOH electrolyte solution, affording a 70% yield of 2a (Table, entry 1). The optimized conditions are the result of combining two alterations discussed in entries 2–5 in Table, where the reaction was limited by the cell voltage reaching the upper limit of the galvanostat used. For this reason, this multifactor interaction was investigated in a design of experiments (DoE) together with the electrochemical parameters and the concentrations of the starting materials (see the Supporting Information for more details). ?,? The DoE provided benzimidazole 2a in 70% yield, which could not be further improved. Using 4 F in sodium acetate (0.27 M) and acetic acid (6 M) in a MeOH electrolyte solution utilizing just 1 equiv of aldehyde affords 52% and 60% yields of 2a (Table, entries 2 and 3, respectively). Increasing the current density results in a 53% yield at 10.0 mA cm^–2^, while decreasing the current density decreases the yield further to 44% (Table, entries 4 and 5). Different cathode materials were screened (Table, entries 6–9).? Lead cathodes afford 2a in 65% yield but were subjected to severe corrosion,? which could not be suppressed. Using diammonium salt additives previously shown to stabilize lead electrodes had no visible effect (see the Supporting Information for details).? Different electrolyte systems were investigated, as well (see the Supporting Information). Of the electrolyte systems tested, a system of acetic acid and sodium acetate in methanol was the only system affording 2a in a sufficient yield while sulfuric acid in methanol was the only other electrolyte to afford molecule 2a, be in a yield of only 5% (Table, entries 1 and 2, and the Supporting Information). An investigation of the reaction temperature indicated that decreasing the reaction temperature had no significant impact on the reaction and afforded molecule 2a in 69% yield. Increasing the temperature decreased the yield of 2a to only 61% (Table, entries 10 and 11). Furthermore, the reaction does not proceed when no electricity is applied (entry 12) or acids/esters are used as condensation partners instead of aldehydes (investigated with 4-fluorobenzoic acid and methyl 4-fluorobenzoate due to the ease of analysis).
Hydrazides 1a–q were synthesized and subjected to the electrochemical protocol (see the Supporting Information for details). The substitution position at the benzo ring made a negligible difference as synthesizing toluoyl hydrazides 2a–c gave yields between 64% and 71%. Benzohydrazide 2d and para-substituted benzohydrazides 2e and 2f were obtained in yields of 62% (R = H), 41% (R = OMe), and 72% (R = ^ t ^Bu), respectively. Different substitution patterns were tested for halo substituents, and 2g–j were obtained in yields between 52% and 67%. Naphthoyl hydrazide 2k was obtained in 58% yield. We were able to isolate alkyl-substituted products 2l and 2m in yields of 52% and 58%, respectively. Phenylacethydrazide 2n was obtained in 42% yield. The method even tolerated a double bond in 1o to yield 2o in 73% isolated yield (see Figure for the structures). However, functional groups that are not stable under reductive conditions (e.g., oxalates) and pyridines, pyrroles, or sterically overly demanding substrates on the aldehyde pose a limitation for this method (see the Supporting Information).
Next, a range of different aldehydes were reacted with 1a to afford the corresponding 2-substituted benzimidazoles. The transformation displayed broad functional group tolerance without compromising the reactivity. p-Methyl-substituted (3a) and halo-substituted (3b–d) compounds, amenable for further postfunctionalization, and electron-withdrawing esters (3e) were obtained in yields ranging from 73% to 78%. Fluorobenzaldehydes substituted at any ring position delivered N-amido benzimidazoles 3f–h in 63–71% yields. The reaction tolerated among other functionalities free phenols at the meta position and m-methoxy and meta-nitrile benzimidazoles (3i–k) were isolated in 72%, 89%, and 81% yields, respectively, with the m-methoxy derivative having the highest overall yield. Additionally, the ortho-substituted methoxy benzaldehyde afforded benzimidazole 3l in 68% yield. Heteroatomic aldehydes, among others, thiophene-2/3-carboxaldehyde and furfural, respectively, 5-methyl furfural, were well tolerated, resulting in N-amido benzimidazoles 3m–p in 68–77% yields. The reaction also tolerated more complex aldehydes like vanillin or (1R)-(−)-myrtenal, resulting in 3q or 3r, respectively, although with slightly lower yields of 58% for both compared to the other used aldehydes. Last, the reaction was performed with formaldehyde and alkyl-substituted aldehydes, affording N-amido benzimidazoles 3s–w with 28% and 40–85% yields, whereas phenylacetaldehyde gave the lowest yield with 40% of the alkyl group-bearing aldehydes (see Figure for structures). A 30-fold scale-up experiment (3.05 g (11.25 mmol) compared to 101.7 mg (0.375 mmol)) was performed using 1a and benzaldehyde (see the Supporting Information) to show the applicability of this method. Desired compound 2a was isolated in 74% yield, in comparison to 65% in the small scale reaction, which could be attributed to more efficient convection and handling on a larger scale.
The structure of molecule 3s was unequivocally confirmed by X-ray single-crystal diffraction and matches the NMR data (see the bottom left of Figure and the Supporting Information).
We established the first electrochemical synthesis to previously difficult-to-access 1,2-aryl-substituted N-amido benzimidazoles under mild conditions in a simple and metal-free key step. The broad applicability of this method was demonstrated with almost 40 examples displaying isolated yields of ≤89%. The method tolerates a myriad of functional groups on both rings of the benzimidazole, including halo substituents, nitriles, alcohols, double bonds, alkyl chains, and even heterocycles at position 2. It represents the first general route to this moiety. The easy access and reliable protocol result in these promising scaffolds with potential biological activity that inherit the potential of postfunctionalization at the amine functionality, opening the door for future pharmaceutical investigations. Furthermore, this electrosynthesis was reliably scaled up.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Vitaku E.Smith D. T.Njardarson J. T.Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals J. Med. Chem.20145724102571027410.1021/jm 501100 b 25255204 · doi ↗ · pubmed ↗
- 2Marshall C. M.Federice J. G.Bell C. N.Cox P. B.Njardarson J. T.An Update on the Nitrogen Heterocycle Compositions and Properties of U.S. FDA-Approved Pharmaceuticals (2013–2023)J. Med. Chem.20246714116221165510.1021/acs.jmedchem.4c 0112238995264 · doi ↗ · pubmed ↗
- 3Brishty S. R.Hossain M. J.Khandaker M. U.Faruque M. R. I.Osman H.Rahman S. M. A.A Comprehensive Account on Recent Progress in Pharmacological Activities of Benzimidazole Derivatives Front. Pharmacol.20211276280710.3389/fphar.2021.76280734803707 PMC 8597275 · doi ↗ · pubmed ↗
- 4Kerru N.Gummidi L.Maddila S.Gangu K. K.Jonnalagadda S. B.A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications Molecules 202025190910.3390/molecules 2508190932326131 PMC 7221918 · doi ↗ · pubmed ↗
- 5Law C. S. W.Yeong K. Y.Benzimidazoles in Drug Discovery: A Patent Review Chem Med Chem.202116121861187710.1002/cmdc.20210000433646618 · doi ↗ · pubmed ↗
- 6Sachs G.Shin J. M.Howden C. W.Review Article: The Clinical Pharmacology of Proton Pump Inhibitors Aliment. Pharmacol. Ther.2006232810.1111/j.1365-2036.2006.02943.x 16700898 · doi ↗ · pubmed ↗
- 7Singh S.Singh N.Kumar V.Datta S.Wani A. B.Singh D.Singh K.Singh J.Toxicity, Monitoring and Biodegradation of the Fungicide Carbendazim Environ. Chem. Lett.201614331732910.1007/s 10311-016-0566-2 · doi ↗
- 8Bansal Y.Silakari O.The Therapeutic Journey of Benzimidazoles: A Review Bioorg. Med. Chem.201220216208623610.1016/j.bmc.2012.09.01323031649 · doi ↗ · pubmed ↗