A trio-binned, haplotype-resolved genome sequence of the zebrafish, Danio rerio Hamilton 1822, SAT strain
Kerstin Howe, Arang Rhie, Sergey Koren, Elisabeth Busch-Nentwich, Shane A. McCarthy, Jonathan M. D. Wood, Michelle Smith, Gene Myers, Karen Oliver, Annabel Whibley, Tim Martin Knutsen, Jayan Duminda M Senevirathna

TL;DR
This paper provides a detailed, haplotype-resolved genome sequence of a zebrafish SAT strain individual.
Contribution
The study presents the first trio-binned, haplotype-resolved genome assemblies for the zebrafish SAT strain.
Findings
The paternal haplotype genome is 1,354.1 megabases long.
The maternal haplotype genome is 1,360.5 megabases long.
The mitochondrial genome is 16.6 kilobases in length.
Abstract
We present the trio-binned, haplotype-resolved genome assemblies (released in 2020) of both haplotypes of an individual of the Danio rerio SAT strain, a cross between Tuebingen (maternal) and AB (paternal) strains (zebrafish; Chordata; Actinopteri; Cypriniformes; Cyprinidae). The genome sequence of the paternal haplotype (fDreABH1) is 1,354.1 megabases long, while the genome sequence of the maternal haplotype (fDreTuH) is 1,360.5 megabases long. Most of the assembly is scaffolded into 25 chromosomal pseudomolecules. The mitochondrial genome has also been assembled and is 16.6 kilobases in length.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
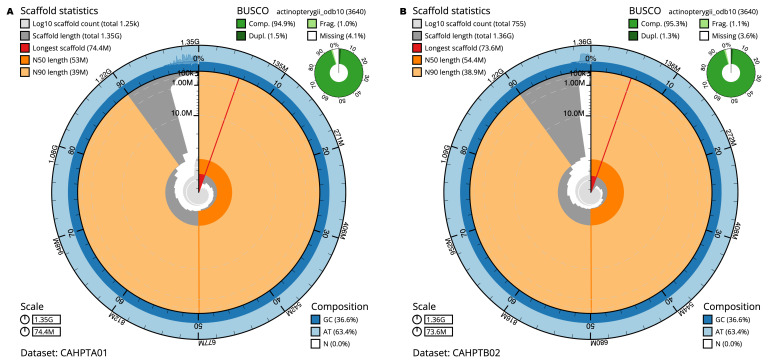 Figure 1
Figure 1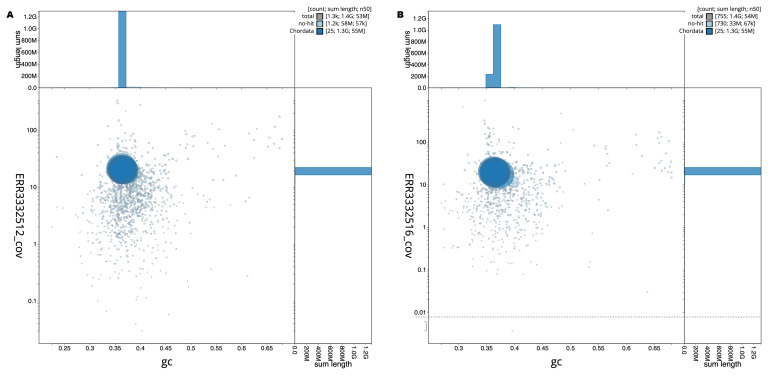 Figure 2
Figure 2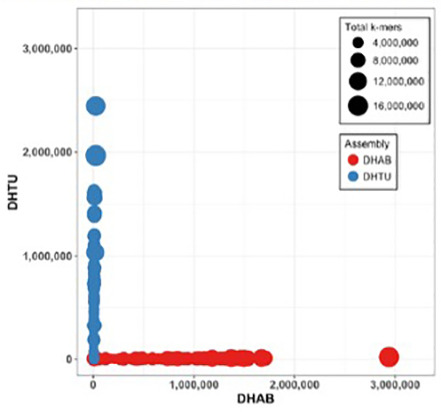 Figure 3
Figure 3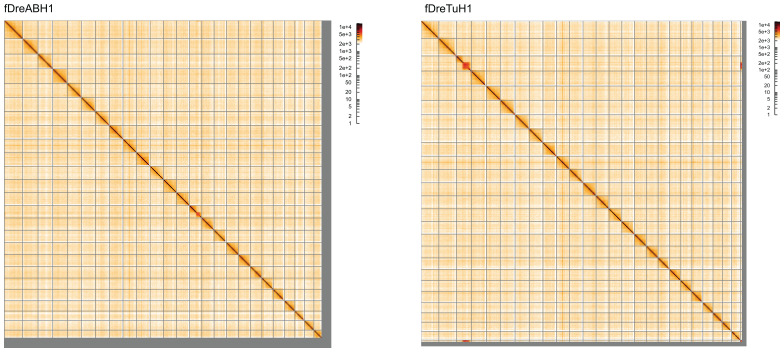 Figure 4
Figure 4| Project accession data | Paternal | Maternal |
|---|---|---|
| Assembly identifiers | fDreABH1.1 | fDreTuH1.2 |
| Species |
| |
| Specimen | fDreABH1 | fDreTuH1 |
| NCBI taxonomy ID | 7955 | |
| BioProject | PRJEB38590 | |
| BioSample ID | SAMEA4760846 | SAMEA4760849 |
| Assembly metrics
| fDreABH1.1 | fDreTuH1.2 |
| BUSCO
| C:94.9%[S:93.3%,D:1.5%],
| C:95.3%[S:94.0%,D:1.3%],
|
| Mitochondrial assembly | - | |
| Raw data accessions | ||
| 10X Genomics Illumina | ERR3332513, ERR3332515, ERR3332516, ERR3332517, ERR3332514, ERR3332511, ERR3332512, ERR3332510, | |
| Hi-C-dovetail | ERR4021773 | |
| Genome assembly | ||
| Assembly accession | GCA_903684865.1 | GCA_903684855.2 |
| Span (Mb) | 1,354.1 | 1,360.5 |
| Number of contigs | 1,964 | 2,104 |
| Contig N50 length (Mb) | 4.5 | 3.6 |
| Number of scaffolds | 1,252 | 754 |
| Scaffold N50 length (Mb) | 53.0 | 54.4 |
| Longest scaffold (Mb) | 74.43 | 73.59 |
| fDreABH1 (paternal haplotype) | fDreTuH1.2 (maternal haplotype) | |||
|---|---|---|---|---|
| Chromosome | INSDC accession | Length (Mb) | INSDC accession | Length (Mb) |
| 1 | 59.96 | 59.57 | ||
| 2 | 57.65 | 59.97 | ||
| 3 | 62.6 | 62.13 | ||
| 4 | 51.23 | 62.64 | ||
| 5 | 74.43 | 72.58 | ||
| 6 | 59.69 | 59.35 | ||
| 7 | 32.28 | 73.59 | ||
| 8 | 55.94 | 55.32 | ||
| 9 | 57.07 | 56.85 | ||
| 10 | 46.41 | 45.64 | ||
| 11 | 46.06 | 45.93 | ||
| 12 | 49.74 | 36.19 | ||
| 13 | 52.96 | 53.21 | ||
| 14 | 54.75 | 53.9 | ||
| 15 | 50.01 | 49.37 | ||
| 16 | 55.93 | 55.35 | ||
| 17 | 54.64 | 54.39 | ||
| 18 | 52.5 | 52.04 | ||
| 19 | 49.85 | 49.53 | ||
| 20 | 56.54 | 55.94 | ||
| 21 | 48.05 | 47.18 | ||
| 22 | 39.02 | 38.89 | ||
| 23 | 47.47 | 47.27 | ||
| 24 | 42.17 | 42.36 | ||
| 25 | 38.62 | 38.18 | ||
| MT | - | - | 0.02 | |
| SATMAP aligns unique | DHAB marker | DHTU marker |
|---|---|---|
|
| 121,208 | 14,882 |
|
| 14,975 | 126,824 |
| AB reads | Tuebingen reads | |
|---|---|---|
|
| 97.55% | 4.59% |
|
| 2.44% | 95.4% |
- —Wellcome Trust
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Genetic diversity and population structure · Zebrafish Biomedical Research Applications
Species taxonomy
Eukaryota; Opisthokonta; Metazoa; Eumetazoa; Bilateria; Deuterostomia; Chordata; Craniata; Vertebrata; Gnathostomata; Teleostomi; Euteleostomi; Actinopterygii; Actinopteri; Neopterygii; Teleostei; Osteoglossocephalai; Clupeocephala; Otomorpha; Ostariophysi; Otophysi; Cypriniphysae; Cypriniformes; Cyprinoidei; Danionidae; Danioninae; Danio; Danio rerio (Hamilton, 1822) (NCBI:txid7955).
Background
Danio rerio, commonly known as the zebrafish, serves as a model organism in various fields of biological research, including genetics, developmental biology, and toxicology ( Lieschke & Currie, 2007). Native to the freshwater habitats of South Asia, the genome of this teleost fish was fully sequenced in 2013 ( Howe et al., 2013). The genome comprises approximately 1.4 gigabases, distributed across 25 chromosomes. Notably, the zebrafish genome exhibits a high degree of synteny with the human genome, making it a valuable tool for studying gene function and regulation ( Howe et al., 2013; Postlethwait et al., 2000).
One of the advantages of using Danio rerio as a model organism is its rapid embryonic development, which is largely transparent and easily observable under a microscope ( Kimmel et al., 1995). This facilitates real-time analysis of developmental processes. Additionally, the zebrafish is amenable to genetic manipulation, including techniques such as CRISPR/Cas9, which allows for targeted gene editing ( Hwang et al., 2013). Its relatively low maintenance cost and high fecundity further contribute to its utility in research settings ( Lawrence, 2007).
The availability of a fully sequenced genome has accelerated functional genomics studies in zebrafish, enabling researchers to perform genome-wide association studies (GWAS), transcriptomics, and other high-throughput analyses ( Lieschke & Currie, 2007). Consequently, Danio rerio continues to be an indispensable resource in advancing our understanding of vertebrate biology, genetics, and disease mechanisms.
In addition to its genomic attributes, the zebrafish model is further enriched by the diversity of its classical laboratory strains. These strains, including AB (ZFIN ID: ZDB-GENO-960809–7), Tuebingen (TU; ZFIN ID: ZDB-GENO-990623–3), and WIK (ZFIN ID: ZDB-GENO-010531–2), have originated from distinct genetic backgrounds. They are a rich source of genetic and phenotypic diversity, which is essential for a broad range of comparative studies ( Brown et al., 2012). This diversity among strains allows for a more comprehensive understanding of genetic influences on various biological processes and phenotypes.
The latest D. rerio reference genome is based on the Tuebingen (TU) strain (GCA_000002035.4). A high-quality de novo genome assembly of the AB strain of Danio rerio has been generated ( Deng et al., 2022), and was compared to the Tuebingen reference genome. Significant structural differences and extensive sequence divergence were identified, including millions of single nucleotide polymorphisms, insertions, deletions, and inversions. These findings highlight the genetic variation within zebrafish strains and underscore the importance of considering these differences in experimental design and comparative studies.
Here we present the genome assembly of an individual of the Danio rerio SAT strain (ZFIN ID: ZDB-GENO-100413-1), a cross between Tuebingen and AB, based on a sample provided by Elisabeth Busch-Nentwich, University of Cambridge.
Genome sequence report
This project provides the genome assemblies of the maternal (Tuebingen) haplotype and the paternal (AB) haplotype of an individual of the Danio rerio SAT strain (ZFIN ID: ZDB-GENO-100413-1), based on a sample provided by Elisabeth Busch-Nentwich, University of Cambridge. The assembly of the haplotypes fDreABH1.1 and fDreTuH1.2 is based on 67× PacBio Sequel II data generated by Gene Myers at the MPI of Molecular Cell Biology and Genetics, Dresden, 96× 10X Genomics Chromium data, BioNano data, and 83x Dovetail Hi-C data (from a different individual, SAMEA104026397, fDreSAT3) generated at the Wellcome Sanger Institute. The trioCanu assembly was created by Arang Rhie at NHGRI, and subsequent steps were carried out at the Sanger Institute. The mitochondrial assembly is provided by the Vertebrate Genomes Project team at The Rockefeller University.
The final fDreABH1.1 assembly has a total length of 1,354.1 Mb in 1,252 sequence scaffolds with a scaffold N50 of 53.0 Mb ( Table 1). The final fDreTuH1.2 assembly has a total length of 1,360.5 Mb in 754 sequence scaffolds with a scaffold N50 of 54.4 Mb ( Table 1). The snail plots in Figure 1 provide a summary of the assembly statistics, while the distribution of assembly scaffolds on GC proportion and coverage is shown in Figure 2.
Table 1.: Genome data for Danio rerio, fDreTuH1.2.
Genome assemblies of Danio rerio SAT: metrics.The BlobToolKit snail plots show N50 metrics and BUSCO gene completeness. The main plot is divided into 1,000 size-ordered bins around the circumference with each bin representing 0.1% of the assembly. The distribution of sequence lengths is shown in dark grey with the plot radius scaled to the longest sequence present in the assembly (shown in red). Orange and pale-orange arcs show the N50 and N90 sequence lengths, respectively. The pale grey spiral shows the cumulative sequence count on a log scale with white scale lines showing successive orders of magnitude. The blue and pale-blue area around the outside of the plot shows the distribution of GC, AT and N percentages in the same bins as the inner plot. A summary of complete, fragmented, duplicated and missing BUSCO genes in the actinopterygii_odb10 set is shown in the top right. The snail plots can be viewed interactively at the following links: fDreABH1 and for fDreTuH1.
Genome assemblies of Danio rerio SAT: BlobToolKit GC-coverage plot.Scaffolds are coloured by phylum. Circles are sized in proportion to scaffold length. Histograms show the distribution of scaffold length sum along each axis. The blob plots can be viewed interactively at the following links: fDreABH1 and for fDreTuH1.
The trio binning approach successfully separated the parental haplotypes based on *k-*mer classification. Haplotype-specific k-mer analysis confirmed that contigs were correctly assigned to their respective parental haplotypes, with minimal cross-contamination between the DHAB (paternal) and DHTU (maternal) assemblies ( Figure 3).
Haplotype k-mer validation for the SAT trio-binned assembly.Each circle represents a contig, with size proportional to total k-mer content. The x-axis shows AB-specific k-mer counts (DHAB: double haploid AB parent), and the y-axis shows Tuebingen-specific k-mer counts (DHTU: double haploid Tuebingen parent). Red circles: contigs assigned to the fDreABH1 (paternal) assembly; blue circles: contigs assigned to the fDreTuH1 (maternal) assembly. Successful phasing is demonstrated by contigs clustering near their respective axes.
Most (95.69%) of the assembly sequence was assigned to 25 chromosomal-level scaffolds. Chromosome-scale scaffolds confirmed by the Hi-C data are named by synteny ( Figure 4; Table 2). The mitochondrial genome was also assembled and can be found as a contig within the multifasta file of the genome submission.
Genome assemblies of Danio rerio SAT: Hi-C contact map of the assemblies, visualised using HiGlass. Chromosomes are shown in order of size from left to right and top to bottom. The panel on the left shows the fDreABH1 assembly and the right show the fDreTuH1 assembly. The maps can be viewed interactively at the following links: fDreABH1 and for fDreTuH1.
Table 2.: Chromosomal pseudomolecules in the genome assembly of Danio rerio, fDreSAT1.
For the maternal haplotype (fDreTuH1.2), the estimated Quality Value (QV) of the final assembly is 35.5 with k-mer completeness of 99.13%, and the assembly has a BUSCO completeness of 95.3% (single = 94.0%, duplicated = 1.3%). BUSCO version 5.3.2 was used, using the actinopterygii_odb10 reference set ( n = 3,640). For the paternal haplotype (fDreABH1.1), the estimated Quality Value (QV) of the final assembly is 36.4 with k-mer completeness of 99.31%, and the assembly has a BUSCO completeness of 94.9% (single = 93.3%, duplicated = 1.5%).
Methods
Sample acquisition and nucleic acid extraction
A sample of an individual of the Danio rerio SAT strain was provided by Elisabeth Busch-Nentwich, University of Cambridge. This strain is a cross between a female of the Tuebingen haplotype and a male of the AB haplotype.
Nucleic acid extraction was carried out using Bionano Prep Cell Culture DNA Isolation Protocol. In this method, the cells are first embedded in agarose to provide structural support during the extraction process. The agarose-embedded cells are then treated with lysis buffers to break down the cell membranes and release the DNA. The process also involves proteinase digestion to remove proteins, followed by a series of washes to purify the DNA.
Sequencing
Pacific Biosciences CLR (continuous long reads) and 10X Genomics read cloud DNA sequencing libraries were constructed according to the manufacturers’ instructions. DNA sequencing was performed by the Scientific Operations core at the WSI on Pacific Biosciences SEQUEL II (CLR) and Illumina HiSeq X Ten instruments. Additionally, BioNano optical maps were generated to provide high-resolution genome mapping. Discovar sequencing of the maternal and paternal samples was conducted on Illumina platforms to improve genome assembly accuracy. Hi-C data were also generated using the Dovetail Hi-C kit and sequenced on the HiSeq X Ten instrument.
Genome assembly, curation and evaluation
The fDreABH1.1 and fDreTuH1.2 haplotype assemblies are based on 67× PacBio CLR data generated by Gene Myers at the MPI of Molecular Cell Biology and Genetics, Dresden, 96× 10X Genomics Chromium data, BioNano data, and 83x Dovetail Hi-C data (from a different individual, SAMEA104026397, fDreSAT3) generated at the Wellcome Sanger Institute. The assembly process included the following sequence of steps: segregation of PacBio reads representing the two haplotypes using trioCanu ( Koren et al., 2018) and k-mers from the Illumina-sequenced parents (maternal: SAMEA3891248 at 143× coverage, and paternal: SAMEA3891249 at 136× coverage), separate assembly of each haplotype using Canu ( Koren et al., 2017), polishing with Arrow from the binned reads, retained duplication removal with purge_dups, 10X based scaffolding with scaff10x, BioNano hybrid-scaffolding, Hi-C based scaffolding with SALSA2 ( Ghurye et al., 2019), further Arrow polishing, and two rounds of FreeBayes ( Garrison & Marth, 2012) Illumina polishing. The mitochondrial assembly was produced by the Vertebrate Genomes Laboratory at Rockefeller University using mitoVGP. Finally, the assembly was analysed and manually improved using gEVAL ( Chow et al., 2016), and marker placement from the high-density genetic map SATmap ( Howe et al., 2013) to achieve chromosomal resolution ( Table 3).
To evaluate the final genome assemblies, the genome was analysed within the BlobToolKit environment ( Challis et al., 2020) and BUSCO scores ( Manni et al., 2021) were calculated. Hi-C maps were produced using bwa-mem2 ( Vasimuddin et al., 2019) in the Cooler file format ( Abdennur & Mirny, 2020).
Alignment of parental Illumina reads to the haplotype assemblies using minimap2 confirmed the correct assignment of sequences, with 97.55% of AB reads mapping to the fDreABH1 assembly and 95.4% of Tuebingen reads mapping to the fDreTuH1 assembly ( Table 4).
Wellcome Sanger Institute – Legal and Governance
The materials that have contributed to this genome note have been supplied by a Tree of Life collaborator. The Wellcome Sanger Institute employs a process whereby due diligence is carried out proportionate to the nature of the materials themselves, and the circumstances under which they have been/are to be collected and provided for use. The purpose of this is to address and mitigate any potential legal and/or ethical implications of receipt and use of the materials as part of the research project, and to ensure that in doing so we align with best practice wherever possible.
The overarching areas of consideration are:
• Ethical review of provenance and sourcing of the material
• Legality of collection, transfer and use (national and international)
Each transfer of samples is undertaken according to a Research Collaboration Agreement or Material Transfer Agreement entered into by the Tree of Life collaborator, Genome Research Limited (operating as the Wellcome Sanger Institute) and in some circumstances other Tree of Life collaborators.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdennur N Mirny LA : Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36(1):311–316. 10.1093/bioinformatics/btz 540 31290943 PMC 8205516 · doi ↗ · pubmed ↗
- 2Brown KH Dobrinski KP Lee AS : Extensive genetic diversity and substructuring among zebrafish strains revealed through copy number variant analysis. Proc Natl Acad Sci U S A. 2012;109(2):529–534. 10.1073/pnas.1112163109 22203992 PMC 3258620 · doi ↗ · pubmed ↗
- 3Challis R Richards E Rajan J : Blob Tool Kit - interactive quality assessment of genome assemblies. G 3 (Bethesda). 2020;10(4):1361–1374. 10.1534/g 3.119.400908 32071071 PMC 7144090 · doi ↗ · pubmed ↗
- 4Chow W Brugger K Caccamo M : g EVAL — a web-based browser for evaluating genome assemblies. Bioinformatics. 2016;32(16):2508–2510. 10.1093/bioinformatics/btw 159 27153597 PMC 4978925 · doi ↗ · pubmed ↗
- 5Deng Y Qian Y Meng M : Extensive sequence divergence between the reference genomes of two zebrafish strains, Tuebingen and AB. Mol Ecol Resour. 2022;22(6):2148–2157. 10.1111/1755-0998.13602 35189030 · doi ↗ · pubmed ↗
- 6Garrison E Marth G : Haplotype-based variant detection from short-read sequencing. 2012. 10.48550/ar Xiv.1207.3907 · doi ↗
- 7Ghurye J Rhie A Walenz BP : Integrating Hi-C links with assembly graphs for chromosome-scale assembly. P Lo S Comput Biol. 2019;15(8): e 1007273. 10.1371/journal.pcbi.1007273 31433799 PMC 6719893 · doi ↗ · pubmed ↗
- 8Howe K Clark MD Torroja CF : The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498–503. 10.1038/nature 12111 23594743 PMC 3703927 · doi ↗ · pubmed ↗