Intradural Extramedullary Thoracic Spinal Paraganglioma in a Young Adult: A Case Report
Serge E Bambule, Tarik Chekrine, Mouna Bourhafour, Medhi Karkouri, Souha Sahraoui

TL;DR
A young adult had a rare spinal tumor removed and recovered well after surgery and radiotherapy.
Contribution
This case report presents a rare instance of intradural extramedullary thoracic spinal paraganglioma in a young adult.
Findings
The patient showed complete recovery of motor deficits after functional rehabilitation.
Radiological follow-up showed no signs of tumor recurrence one year post-radiotherapy.
Abstract
Spinal paragangliomas are rare, typically benign neuroendocrine tumors, exceptionally uncommon in the intradural extramedullary thoracic region. Their diagnosis is particularly unusual in young adults. We report the case of a 27-year-old patient who presented with progressive paraparesis due to spinal cord compression caused by a 36 × 17 mm mass at the D3-D4 level, identified on MRI. Surgical resection was performed. Pathological examination confirmed the diagnosis of spinal paraganglioma, supported by immunohistochemistry (positive for chromogranin A, synaptophysin, and focal expression of S-100 protein). Given the risk of local recurrence, adjuvant radiotherapy was administered. Clinical outcome was favorable, with complete recovery of motor deficits following functional rehabilitation. Radiological follow-up at one year post-radiotherapy showed no signs of recurrence.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
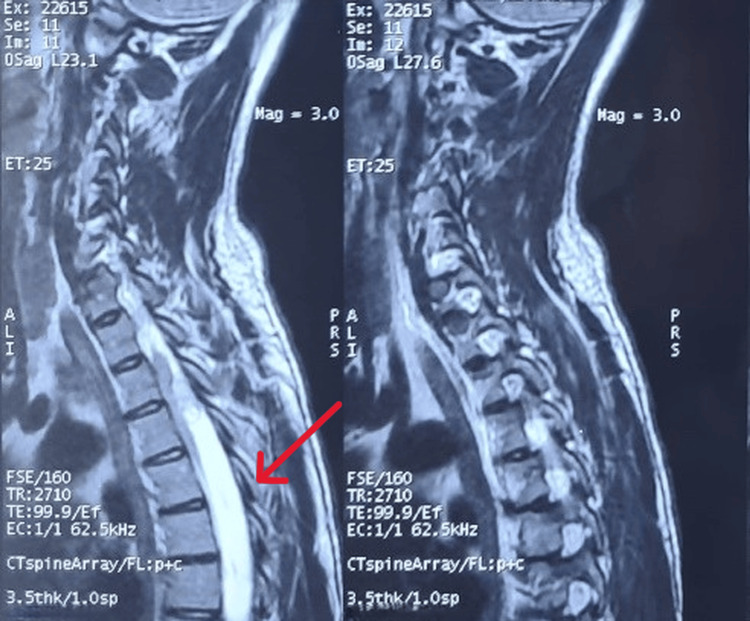 Figure 1
Figure 1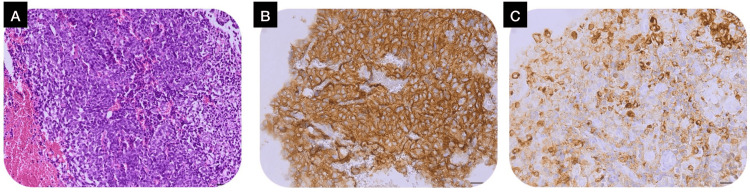 Figure 2
Figure 2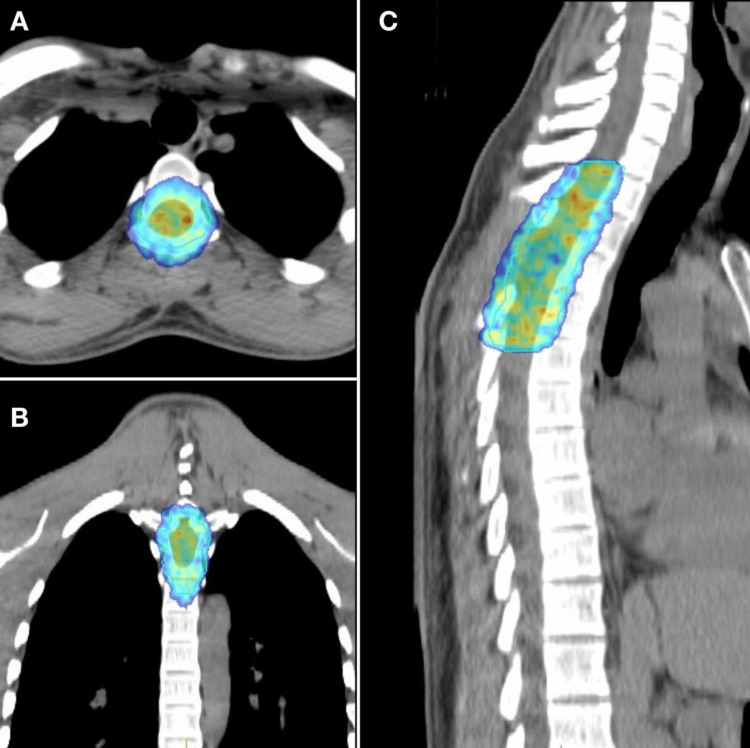 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdrenal and Paraganglionic Tumors · Pituitary Gland Disorders and Treatments · Neurofibromatosis and Schwannoma Cases
Introduction
Paragangliomas are rare neuroendocrine tumors with an estimated global incidence of 2 to 8 cases per million [1]. No specific data on African continental incidence are available; African data are extrapolated from global figures. These tumors originate from paraganglion cells derived from the neural crest and develop along vasculo-nervous axes [1]. Approximately 70% are benign, with malignancy defined by the presence of metastases [2]. At the spinal level, the classic form is intradural extramedullary, predominantly affecting the filum terminale and cauda equina [1,3]. Thoracic or cervical locations are far rarer and may radiologically mimic meningiomas or schwannomas, making preoperative diagnosis challenging [4,5]. Confirmation relies on histopathological examination, often supplemented by immunohistochemistry. Treatment is based on complete surgical resection, the primary objective, and may be complemented by adjuvant radiotherapy to reduce the risk of recurrence [6].
We report a rare case of thoracic spinal paraganglioma in a young adult, highlighting the rarity of this location, diagnostic challenges, and the importance of multidisciplinary management combining surgery and radiotherapy.
Case presentation
Patient information
A 27-year-old man with no significant medical history presented with progressive lower limb weakness, evolving over three months into paraparesis. There were no sensory disturbances, sphincter dysfunction, or systemic symptoms. General condition was preserved.
Clinical findings
Neurological examination revealed bilateral lower-limb weakness graded 3/5 on the Medical Research Council scale. Sensory examination, deep tendon reflexes, and sphincter function were normal.
Timeline
In December 2023, spinal MRI identified an intradural extramedullary mass at D3-D4. Surgical resection was performed. In June 2024, the postoperative MRI showed persistent poorly defined subarachnoid infiltration from D3 to D8 with hyper signal, suggesting residual disease. 18-fluorodeoxyglucose (FDG) PET demonstrated no hypermetabolic lesions. In July-August 2024, adjuvant radiotherapy was delivered, and in August 2025, follow-up MRI confirmed absence of recurrence.
Diagnostic assessment
MRI demonstrated a well-defined intradural extramedullary lesion at the D3-D4 level measuring 36 × 17 mm, causing significant spinal cord compression (Figure 1). Histopathological examination revealed a hemorrhagic tumor proliferation arranged in layers and focally in clusters. Immunohistochemistry showed diffuse positivity for chromogranin A and synaptophysin, with focal S-100 protein expression, confirming the diagnosis of paraganglioma despite the atypical histological appearance (Figure 2). Plasma free metanephrines and normetanephrines were within normal limits, consistent with a nonfunctional tumor.
Preoperative thoracic spine MRI (T2-weighted fast spin-echo (FSE) sequence) showing a 36 × 17 mm intradural extramedullary mass at the D3–D4 level, associated with a focal intramedullary T2 hyperintensity indicating spinal cord involvement.
Histopathological samples from paraganglioma: A) Hemorrhagic tumor proliferation arranged in layers and focally in clusters, hematoxylin-eosin stain, ×20, scale bar = 50 microns. B) Positive expression of synaptophysin, hematoxylin-eosin stain, ×40, scale bar = 50 microns. C) Positive expression of chromogranin A, hematoxylin-eosin stain, ×40, scale bar = 50 microns.
Diagnosis
Diagnosis was a nonfunctional intradural extramedullary thoracic spinal paraganglioma.
Therapeutic interventions
Surgical excision of the tumor was achieved, followed by functional rehabilitation. Given the persistence of poorly defined subarachnoid infiltration with hypersignal suggesting residual disease on postoperative MRI, adjuvant external beam radiotherapy was administered using volumetric modulated arc therapy (VMAT), targeting the D1-D4 region with a total dose of 45 Gy delivered in 1.8 Gy fractions (Figure 3).
Volumetric modulated arc therapy (VMAT) radiotherapy to the D1–D4 region, shown on (A) axial, (B) coronal, and (C) sagittal views.
Follow-up and outcomes
The patient experienced complete recovery of motor function. At 12-month follow-up post-radiotherapy, clinical examination was normal and spinal MRI showed no evidence of local recurrence.
Patient perspective
The patient reported satisfaction with the outcome and relief after regaining full mobility, acknowledging the importance of continued follow-up. Informed consent was obtained.
Discussion
Intradural extramedullary spinal paragangliomas, though rare, can occur at any age. The typical age of onset is between 30 and 50 years, with a peak around 40 years for sporadic cases [1,2]. Hereditary forms, associated with genetic syndromes, may present earlier, often between 20 and 30 years [1,2]. Approximately 30-40% are linked to genetic forms, particularly mutations in SDHB, SDHC, SDHD, VHL, RET, and other genes [1,2]. This prevalence underscores the importance of genetic screening, especially in young patients, those with multiple tumors, or those with a family history. In the African context, limited access to advanced imaging and genetic testing may contribute to underdiagnosis, explaining the scarcity of reported cases in local literature. Genetic testing was not performed in our patient.
Clinically, these tumors typically present with progressive spinal cord compression, without adrenergic symptoms in nonfunctional forms [1,4,5]. Our patient exhibited a similar presentation without headaches, palpitations, or hypertension, confirmed by negative biological tests. Most spinal paragangliomas are non-secreting [1]. The limited data available in the literature confirms the rarity of this location, describing clinical features dominated by spinal cord compression or localized pain, without adrenergic signs [4,5]. Histologically, these tumors are characterized by a nest-like or cord-like architecture of tumor cells (zellballen pattern), surrounded by sustentacular cells and a rich vascular network [1]. Paragangliomas express specific neuroendocrine markers, such as chromogranin A and synaptophysin, used to confirm the diagnosis, while sustentacular cells express S-100 protein, a key marker for their identification. Additional markers, such as tyrosine hydroxylase or dopamine beta-hydroxylase, reflect their chromaffin cell origin [1]. In our case, despite the absence of a typical morphological pattern, immunohistochemistry confirmed the diagnosis. Functional diagnosis relies on measuring catecholamine metabolites, particularly plasma or 24-hour urinary metanephrines and normetanephrines [2]. Certain medications, such as tricyclic antidepressants or antipsychotics, can elevate these levels and lead to false positives [1]. Plasma chromogranin A, though used as a neuroendocrine marker, is less sensitive and specific than catecholamine metabolites [2]. In our patient, no preoperative adrenergic signs were noted, and postoperative biological tests were negative. Radiologically, diagnosis involves conventional and functional imaging. On spinal MRI, paragangliomas typically appear as well-defined, highly vascularized masses with marked contrast enhancement, sometimes associated with hemorrhagic or cystic areas. These features are non-specific, necessitating differentiation from meningiomas, schwannomas, or other hypervascular lesions [1]. Nuclear imaging is valuable for detecting multifocal or metastatic lesions [7]. Several imaging techniques are available, including metaiodobenzylguanidine (MIBG) scintigraphy, 18-FDG PET, 18 F-Dopa PET, and, in some cases, octreotide scintigraphy, although newer tracers like 68Ga-DOTATATE offer increased sensitivity for metastatic lesions [7]. Surgery is the cornerstone of treatment. Complete resection is critical to minimize recurrence risk [5,8,9]. Radiotherapy is indicated postoperatively for incomplete resection or aggressive tumors [8,10]. For benign spinal paragangliomas, stereotactic radiotherapy is also used for unresectable tumors or recurrences [6]. In conventional or intensity-modulated radiotherapy (IMRT), doses typically range from 45 Gy to 50-54 Gy for residual volumes, delivered in 1.8-2 Gy daily fractions [10]. Stereotactic radiotherapy provides symptomatic improvement in 85-87% of patients and rare toxicity (<1% myelopathy) for doses of 14-30 Gy in one to five fractions, offering long-term tumor control (92% at five years, 90% at 10 years) [11]. In our case, the patient received adjuvant radiotherapy of 45 Gy in 1.8 Gy fractions to the D1-D4 region. This case is unusual due to the exceptional intradural extramedullary thoracic location of the paraganglioma in a 27-year-old patient. Postoperative MRI revealed a diffuse and persistent subarachnoid hypersignal, suggestive of tumor residue, with radiologists considering it unlikely that this was solely due to postoperative changes. After multidisciplinary discussion, adjuvant radiotherapy was decided upon to reduce the risk of recurrence. Histological confirmation was not performed for this infiltration.
Lifelong surveillance is recommended. The risk of local or distant recurrence is estimated at approximately 10% at five to 10 years, particularly in patients with genetic forms [1]. This case adds to the limited literature on thoracic spinal paragangliomas in young adults and supports the role of multidisciplinary management.
Conclusions
Thoracic intradural extramedullary spinal paragangliomas are exceptionally rare, especially in young adults. Accurate diagnosis requires correlation of imaging, histopathology, and immunohistochemistry. Complete surgical resection remains the cornerstone of treatment, while adjuvant radiotherapy may provide additional local control in selected cases. Long-term surveillance is essential to detect recurrence.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pheochromocytoma and paraganglioma N Engl J Med Neumann HP Young WF Jr Eng C 55256538120193139050110.1056/NEJ Mra 1806651 · doi ↗ · pubmed ↗
- 2[Phaeochromocytoma and paraganglioma]Rev Med Interne Cornu E Belmihoub I Burnichon N 7337414020193149393810.1016/j.revmed.2019.07.008 · doi ↗ · pubmed ↗
- 3Paragangliomas of the lumbar region. Report of two cases and review of the literature J Neurosurg Spine Gelabert-González M 354365220051579636310.3171/spi.2005.2.3.0354 · doi ↗ · pubmed ↗
- 4MR findings of the spinal paraganglioma: report of three cases J Korean Med Sci Shin JY Lee SM Hwang MY Sohn CH Suh SJ 5225261620011151180310.3346/jkms.2001.16.4.522PMC 3054770 · doi ↗ · pubmed ↗
- 5Cervical intradural paraganglioma presenting as progressive cervicodynia: case report and literature review Clin Neurol Neurosurg Lu L Dai Z Zhong Y Lv G 35936111520132272177410.1016/j.clineuro.2012.05.029 · doi ↗ · pubmed ↗
- 6Treatment for paraganglioma with stereotactic radiotherapy World J Clin Cases Pontoriero A Critelli P Zeppieri M Angileri FF Ius T 272927371220243889928910.12998/wjcc.v 12.i 16.2729 PMC 11185345 · doi ↗ · pubmed ↗
- 7PET/CT comparing (68)Ga-DOTATATE and other radiopharmaceuticals and in comparison with CT/MRI for the localization of sporadic metastatic pheochromocytoma and paraganglioma Eur J Nucl Med Mol Imaging Janssen I Chen CC Millo CM 178417914320162699677910.1007/s 00259-016-3357-x PMC 8194362 · doi ↗ · pubmed ↗
- 8Clinical characteristics and surgical treatment of spinal paraganglioma: a case series of 18 patients Clin Neurol Neurosurg Yin M Huan Q Sun Z 202615820172843372510.1016/j.clineuro.2017.03.019 · doi ↗ · pubmed ↗