Episodic Ataxia Associated With Synaptosomal-Associated Protein 25 (SNAP25) Variant: Beyond Epilepsy and Developmental Delay
Inês F Fernandes, Ana Cristina Figueiredo, João Parente Freixo, Juliette Dupont, João Carvalho

TL;DR
A child with a new SNAP25 gene variant showed seizures, developmental delay, and febrile-triggered ataxia, expanding the known effects of this genetic condition.
Contribution
A novel de novo SNAP25 splice-site variant is reported, adding febrile-triggered episodic ataxia to the phenotypic spectrum of SNAP25-related disorders.
Findings
A novel SNAP25 variant (c.114+2dup) was identified in a child with seizures and developmental delay.
Febrile illness-triggered episodic ataxia is a newly reported feature of SNAP25-related developmental and epileptic encephalopathy.
Levetiracetam monotherapy achieved complete seizure control in the patient.
Abstract
Developmental and epileptic encephalopathies (DEEs) of infancy and childhood are characterized by early-onset seizures and developmental impairment. Heterozygous missense or loss-of-function variants in synaptosomal-associated protein 25 (SNAP25), a core component of the presynaptic soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, have been implicated in a spectrum of DEEs with variable neurological features. We report a male child carrying a novel de novo splice-site variant in SNAP25, expanding the known mutational and phenotypic spectrum of this condition. The patient presented at 32 months with afebrile seizures since seven months of age, with frequent seizure clusters and status epilepticus. He exhibited a moderate global developmental delay and recurrent, transient episodes of gait ataxia triggered by febrile illnesses, lasting up to one week…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
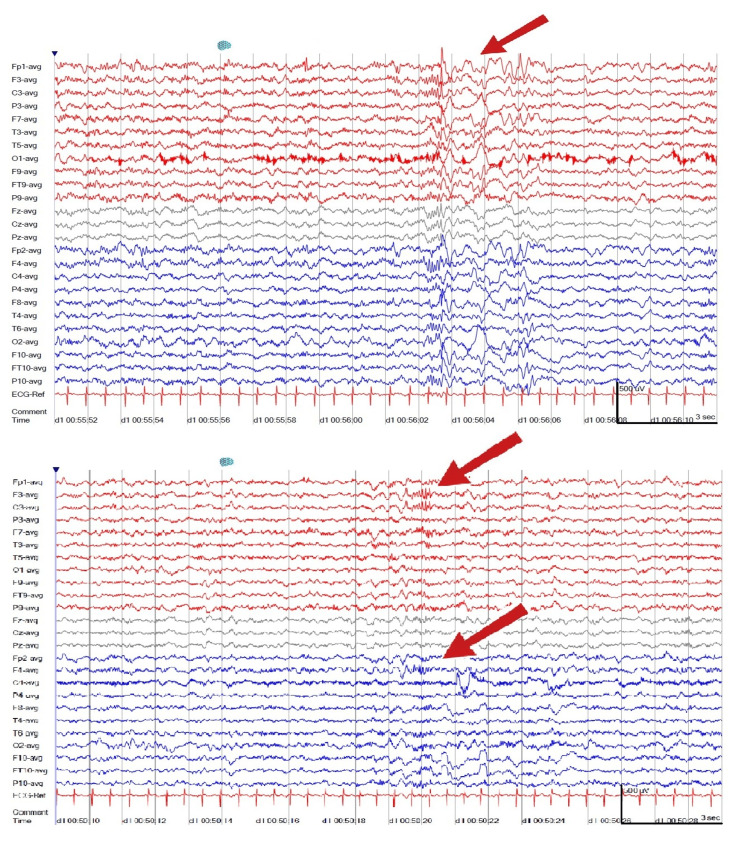 Figure 1
Figure 1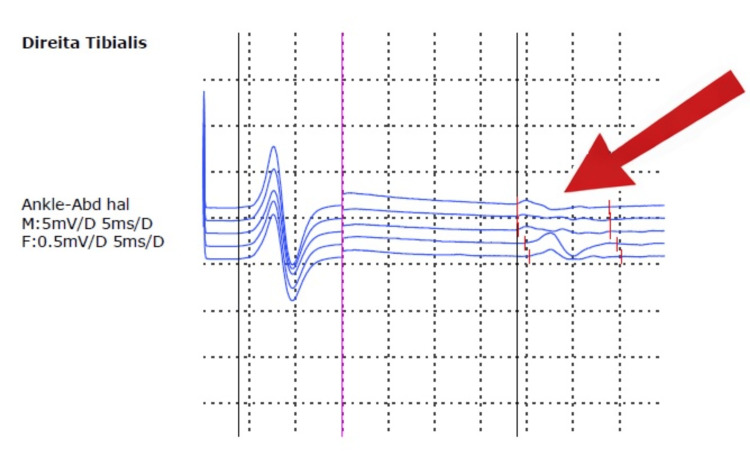 Figure 2
Figure 2| Investigation | Findings | Interpretation |
| Brain MRI | Normal | No structural abnormalities identified |
| EEG | Abundant bilateral frontal paroxysmal activity | Consistent with epileptic encephalopathy |
| Expanded metabolic panel | Plasma amino acids; CSF amino acids; blood lactate & pyruvate; biotinidase; CDT profile; 24-h urine creatine metabolism: all within normal limits | No evidence of inborn metabolic disorder |
| Acylcarnitine profile | Transient elevation of 3-hydroxy-isovalerylcarnitine | Considered valproate-related |
| Urine organic acids | Transient orotic aciduria | Considered valproate-related |
| Electromyography | Myopathic motor unit potentials in right tibialis anterior; normal nerve conduction velocities; normal repetitive nerve stimulation | No neuromuscular junction dysfunction |
| Genetic testing | Heterozygous | Likely pathogenic |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCellular transport and secretion · Neuroscience and Neuropharmacology Research · Epilepsy research and treatment
Introduction
Developmental and epileptic encephalopathies (DEE) of infancy and childhood are a group of heterogeneous and treatment-resistant disorders characterized by developmental slowing as a direct result of epileptic activity, the underlying etiology, or a combination of both [1,2]. Over 900 genes and structural causes are linked to DEEs [1].
Soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, composed of synaptobrevin, syntaxin, and synaptosomal-associated protein 25 (SNAP25), forms the essential fusion machinery for neurotransmitter release, through synaptic vesicle exocytosis [3]. Recent studies have identified mutations in the gene encoding SNAP25 as causative factors for DEEs of infancy and childhood, associated with diverse clinical manifestations [2,3]. To date, 26 pathogenic or likely pathogenic variants have been reported in the Genome Aggregation Database (gnomAD).
The most consistent and well-established clinical features of SNAP25 developmental and epileptic encephalopathy (SNAP25-DEE) include developmental delay, intellectual impairment, and early-onset seizures, typically beginning before two years of age [4]. Additional neurological manifestations include muscular hypotonia, movement disorders (ataxia, dystonia, or tremor), cerebral visual impairment, and brain volume loss, but these are less universally present and show considerable interindividual expressivity [4]. Phenotypic expression is highly heterogeneous, particularly in terms of seizure type, treatment response, severity of neurodevelopmental impairment, and associated cerebellar ataxia or dysmorphic features [2]. The concept of "SNAREopathies" highlights overlapping phenotypes among genes encoding SNARE complex proteins; however, precise genotype-phenotype mapping within SNAP25-related disease remains incompletely characterized [4].
Case presentation
We report a male child born to non-consanguineous parents. The father had bilateral ptosis without fatigability, ophthalmoparesis, weakness, or ataxia. The mother, three siblings, and one paternal half-brother had no relevant medical history. The maternal family history was notable for three cousins with childhood-onset epilepsy but no impaired neurodevelopment.
Pregnancy was achieved by in vitro fertilization and was uneventful. Delivery occurred at 39 weeks by caesarean section, with Apgar scores of 9/10/10. Birth measurements and the neonatal period were unremarkable.
The child was healthy until seven months of age, when he developed clusters of seizures and status epilepticus in an afebrile context. Seizure semiology included alternating unilateral clonic seizures, generalized tonic-clonic seizures (some beginning with a barking-like vocalization), and brief behavioral arrest without ocular deviation. He eventually developed moderate global developmental delay (without regression), along with a developmental motor coordination disorder. Since the age of two years, febrile illnesses have triggered transient episodes of gait ataxia with no focal weakness, lasting up to one week.
On examination, no dysmorphic features were observed, though mild microcephaly (≈ -2 standard deviation, Nellhaus scale) was noted. Neurological evaluation between episodes of illness revealed no frank gait ataxia, but slight intoeing, foot dragging, and reduced hip elevation during gait. Muscle tone and deep tendon reflexes were normal. The patient was initially treated in his country of origin with phenobarbital (6 mg/kg/day), clonazepam (0,05mg/kg/day), and valproate (~40 mg/kg/day), with incomplete seizure control and mild elevation of transaminases and gamma-glutamyltransferase. At age 32 months, when he first came to our center’s neurology clinic, he was gradually switched to levetiracetam, achieving seizure freedom with high doses.
Brain magnetic resonance imaging (MRI) was normal. An electroencephalogram (EEG) performed at two years and five months showed abundant bilateral frontal paroxysmal activity (Figure 1). Metabolic investigations were unremarkable, except for transient elevation of 3-hydroxy-isovalerilcarnitine and orotic aciduria, considered valproate-related. Electromyography (EMG) demonstrated myopathic motor unit potentials in the right tibialis anterior, with normal nerve conduction velocities and repetitive nerve stimulation (Figure 2). Given the father’s personal history, he also underwent EMG, which showed borderline myopathic findings in the biceps brachii but no evidence of neuromuscular junction dysfunction. The diagnostic workup is summarized in Table 1.
Electroencephalogram showing bilateral frontal epileptiform activityInterictal EEG demonstrates abundant bilateral frontal paroxysmal discharges (arrows), consistent with epileptiform activity. Recording performed at 2 years and 5 months of age.
Electromyography of the right tibialis anterior muscleRepresentative motor unit potentials (arrow) show low-amplitude, short-duration, and simplified morphology, consistent with a myopathic pattern. Nerve conduction velocities and repetitive nerve stimulation were normal (not shown).
A whole-exome-based multigene epilepsy panel identified a heterozygous variant of uncertain significance in SNAP25 (NM_003081.5:c.114+2dup). Subsequent parental analysis demonstrated a de novo origin. The variant affects the canonical +2 donor splice site, and in silico analyses predict disruption of normal splicing. The variant is absent from population databases (gnomAD v4.1), and the patient’s clinical phenotype is highly concordant with SNAP25-DEE. Based on these findings, the variant was reclassified as likely pathogenic according to American College of Medical Genetics and Genomics (ACMG)/ Association for Molecular Pathology (AMP) criteria. Functional validation of aberrant splicing has not yet been performed.
The patient is currently on levetiracetam monotherapy (60 mg/kg/day), with complete seizure control. He has also shown gradual improvement in episodic imbalance and weakness, as well as steady progress in neurodevelopment with ongoing supportive therapies. His expressive language has significantly advanced - he is now able to produce and combine multiple words with good intelligibility. He demonstrates age-appropriate social engagement, naming skills, playfulness, and interaction with peers.
Discussion
Klöckner et al. [4] described a cohort of 23 individuals with de novo SNAP25 variants, revealing a broad phenotypic spectrum. All presented with developmental delay and varying degrees of intellectual disability and motor impairment. Regression occurred in 29% of cases, some coinciding with seizure onset, which was not observed in our patient. Seizures were reported in 74% of patients, with a median onset at 12 months, encompassing epileptic spasms, generalized and focal seizures, and multiple seizure types over time [4], consistent with our case. Half of the individuals were treated with more than three antiepileptic drugs and still had frequent seizures [4]. Our patient had difficult seizure control initially, but is now on monotherapy with levetiracetam.
Muscular hypotonia, one of the most frequent findings in the cohort, was not described in our patient. It is important to note that we first evaluated him at age 32 months, so earlier hypotonia cannot be ruled out. Although EMG showed myopathic motor unit potentials, he exhibited no weakness, even during febrile illness-related exacerbations. Additional findings included movement disorders such as dystonia, tremor, and ataxia (reported in 33% of cases). None of the patients with ataxia were described as having episodic exacerbations during infections [4]. To our knowledge, there are no prior reports of episodic ataxia or other paroxysmal movement disorders, particularly occurring during febrile illnesses, as seen in our case.
Episodic ataxia is most frequently associated with calcium voltage-gated channel subunit alpha1 A (CACNA1A) defects, particularly episodic ataxia type 2, and is defined by recurrent, transient episodes of cerebellar dysfunction (ataxia, vertigo, dysarthria) that may be triggered by fever, stress, or exertion, with interictal periods of normal or near-normal neurological function [5]. CACNA1A encodes the α1A subunit of the P/Q-type voltage-gated calcium channel, which is essential for presynaptic calcium influx and neurotransmitter release [5]. SNAP25 is a key SNARE protein in the same pathway, modulating calcium channel function and vesicle fusion [2,6]. Functional and structural studies of SNAP25 variants demonstrate disruption of SNARE complex assembly and synaptic transmission, either by impairing protein interactions or destabilizing the complex, leading to defective neurotransmitter release [4]. Although these proteins act at distinct molecular steps, they converge functionally on presynaptic release mechanisms.
Episodic ataxia is most commonly associated with CACNA1A and potassium voltage-gated channel subfamily A member 1 (KCNA1) dysfunction, but may also occur in other genetic epilepsies and metabolic disorders [7]. Proline-rich transmembrane protein 2 (PRRT2) variants are implicated in paroxysmal movement disorders [7,8], particularly paroxysmal kinesigenic dyskinesia, which is characterized by sudden and brief attacks of choreoathetosis or dystonia. Xu et al. [8] described an interaction between PRRT2 and presynaptic proteins, including SNARE components such as SNAP25, suggesting PRRT2 may influence synaptic transmission and contribute to paroxysmal phenotypes through modulation of presynaptic machinery. Severe phenotypes, including episodic ataxia, intellectual disability, and infantile seizures, have been reported in patients with biallelic PRRT2 pathogenic variants [9,10].
Based on this functional convergence, we propose the hypothesis that disruption of synaptic transmission caused by a SNAP25 variant may contribute to the transient, illness-associated ataxia observed in our patient, through mechanisms parallel to those seen in CACNA1A and PRRT2-related disorders, although episodic ataxia is not yet a well-established phenotype of SNAP25-DEE in the literature.
Most individuals with SNAP25-DEE do not present behavioral disturbances [4], in line with our patient’s normal social interaction. Microcephaly has been documented in a minority of cases [3,11] of SNAP25-associated disease.
To our knowledge, this is the first report of recurrent episodic ataxia triggered by febrile illness in SNAP25-associated disease. Strengths of this report include detailed longitudinal characterization, integration of neurophysiological and genetic findings, and the identification of a previously unreported de novo splice-site variant. Limitations include the absence of functional studies to confirm the predicted splicing effect.
Conclusions
This case highlights the importance of considering SNAP25 variants in infants with early-onset seizures, developmental delay, and episodic neurological decompensations. We report a novel de novo splice-site variant in SNAP25 associated with a clinical phenotype that includes febrile illness-triggered episodic ataxia, representing a candidate phenotypic association that warrants further investigation. Importantly, our findings emphasize the potential for seizure control and neurodevelopmental improvement under tailored therapy, despite persistent neurological vulnerability.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Operational definition of developmental and epileptic encephalopathies to underpin the design of therapeutic trials Epilepsia Scheffer IE French J Valente KD Auvin S Cross JH Specchio N 101410236620254001391410.1111/epi.18265 PMC 11997937 · doi ↗ · pubmed ↗
- 2Role of aberrant spontaneous neurotransmission in SNAP 25-associated encephalopathies Neuron Alten B Zhou Q Shin OH 597210920213314744210.1016/j.neuron.2020.10.012PMC 7790958 · doi ↗ · pubmed ↗
- 3SNAR Eopathies: diversity in mechanisms and symptoms Neuron Verhage M Sørensen JB 223710720203255941610.1016/j.neuron.2020.05.036 · doi ↗ · pubmed ↗
- 4De novo variants in SNAP 25 cause an early-onset developmental and epileptic encephalopathy Genet Med Klöckner C Sticht H Zacher P 6536602320213329914610.1038/s 41436-020-01020-w · doi ↗ · pubmed ↗
- 5Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS Nat Rev Neurol Rajakulendran S Kaski D Hanna MG 8696820122224983910.1038/nrneurol.2011.228 · doi ↗ · pubmed ↗
- 6The control of neuronal calcium homeostasis by SNAP-25 and its impact on neurotransmitter release Neuroscience Pozzi D Corradini I Matteoli M 72784202018 https://www.sciencedirect.com/science/article/abs/pii/S 03064522183073343047652710.1016/j.neuroscience.2018.11.009 · doi ↗ · pubmed ↗
- 7Episodic ataxias: primary and secondary etiologies, treatment, and classification approaches Tremor Other Hyperkinet Mov (N Y) Hassan A 91320233700899310.5334/tohm.747PMC 10064912 · doi ↗ · pubmed ↗
- 8Paroxysmal kinesigenic dyskinesia: genetics and pathophysiological mechanisms Neurosci Bull Xu JJ Li HF Wu ZY 9529624020243809124410.1007/s 12264-023-01157-z PMC 11250761 · doi ↗ · pubmed ↗