Orbital Rhabdomyosarcoma in a Young Adult With Prior Childhood Leukaemia: A Case Report
Ling Paulina Gronczewska, Osman Haji, Raza A Syed, Ibrahim Basar

TL;DR
A young adult with a history of childhood leukemia developed a rare aggressive tumor in the eye socket, highlighting diagnostic and treatment challenges.
Contribution
This case report adds to the limited literature on orbital RMS in young adults with prior cancer history.
Findings
Orbital RMS is rare in young adults and can follow prior cancer treatment.
Early imaging and biopsy are crucial for diagnosis.
Managing RMS in patients with prior oncological history is complex.
Abstract
Rhabdomyosarcoma (RMS) is a rare and aggressive soft tissue malignancy primarily affecting children and adolescents. Orbital involvement is an uncommon presentation, particularly in young adults. In this case, we present a 21-year-old male with a history of childhood leukaemia treated with chemotherapy and bone marrow transplantation, who presented with a rapidly growing medial orbital mass subsequently diagnosed as RMS. The case emphasises the difficulties in diagnosing the condition, the value of early imaging and biopsy, and the complexities in managing orbital RMS in a patient with a significant oncological history.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
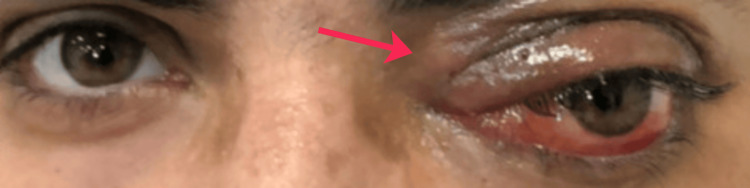 Figure 1
Figure 1| Parameter | Result | Reference Range/Normal Values | Comment |
| Best Corrected Visual Acuity | Right eye: 6/4.8, Left eye: 6/24-1 | 6/6 (20/20) for each eye | Decreased visual acuity in the left eye |
| Colour Vision Testing | Right eye: 15/15, Left eye: 9/15 | 15/15 for each eye | Decreased colour vision in the left eye |
| Intraocular Pressure (IOP) | Right eye: 18 mmHg, Left eye: 38 mmHg | 10-21 mmHg for each eye | Elevated IOP in the left eye |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSarcoma Diagnosis and Treatment · Bone Tumor Diagnosis and Treatments · Vascular Tumors and Angiosarcomas
Introduction
Rhabdomyosarcoma (RMS) is an aggressive malignant soft-tissue tumour arising from skeletal muscle progenitors and represents the most common soft-tissue sarcoma of the head and neck in childhood, with approximately 10% of cases involving the orbit [1,2]. Although predominantly a paediatric malignancy, RMS is rare in adults and is associated with poorer outcomes compared with childhood disease [3]. Because of its aggressive nature and propensity to cause rapid visual and structural deterioration, early recognition and prompt multidisciplinary management are essential.
RMS comprises several histological subtypes, most commonly embryonal and alveolar variants, with embryonal tumours predominating in the head and neck region. Orbital disease typically presents with rapidly progressive proptosis, eyelid or medial canthal swelling, diplopia, raised intraocular pressure, and potential optic nerve compromise - features that can closely mimic more common infectious or inflammatory orbital disorders encountered in general practice [4,5]. These overlapping presentations may delay diagnosis unless clinicians maintain a high index of suspicion.
Survivors of childhood leukaemia treated with intensive chemotherapy, radiotherapy, or stem-cell transplantation are recognised to be at increased risk of therapy-related secondary malignancies due to treatment-induced mutagenic effects on developing tissues. Such secondary cancers may include haematological malignancies and solid tumours, including sarcomas, emerging years after the initial therapy.
We report a rare case of adult-onset orbital RMS in a young adult with a history of childhood leukaemia treated with chemotherapy and bone marrow transplantation, highlighting its atypical presentation, potential therapy-related aetiology, and the diagnostic and management challenges for non-specialist clinicians.
Case presentation
History
A 21-year-old male presented to the ophthalmology clinic at a District General Hospital on February 17, 2025, with a two-week history of swelling in the left medial canthal area, associated with intermittent fever and recent coryzal symptoms. He reported progressive diplopia, particularly on lateral gaze. There was no history of trauma or visual loss.
His past medical history was significant for acute leukaemia diagnosed at the age of four, for which he was treated with chemotherapy and underwent two bone marrow transplants. He also had a history of bilateral avascular necrosis of the hips, requiring hip replacements.
Examination
On inspection and palpation, there was a tender, firm mass in the left medial canthal region associated with proptosis and inferolateral displacement of the globe (Figure 1). Extraocular movements were restricted in all directions in the left eye.
Clinical photograph demonstrating a firm medial canthal mass (arrow), associated left-sided proptosis, and inferolateral displacement of the globe
Best-corrected visual acuity was 6/4.8 in the right eye and 6/24-1 in the left. Colour vision testing revealed 15/15 in the right eye and 9/15 in the left. Intraocular pressure measured 18 mmHg in the right eye and 38 mmHg in the left. Marked chemosis and punctate epithelial changes were present in the left eye. No relative afferent pupillary defect was detected (Table 1).
Investigations
Given the acute presentation with concern for infectious or inflammatory pathology, and the need to rapidly assess bony involvement of the medial orbital wall and paranasal sinuses, a contrast-enhanced CT was selected as the first-line imaging modality. The contrast-enhanced CT scan of the orbit was performed on February 18, 2025, which demonstrated a soft tissue mass originating from the ethmoid sinus with erosion through the medial orbital wall. The lesion extended into the orbit, exerting mass effect on the globe and extending posteriorly towards the orbital apex.
Although the patient initially reported coryzal symptoms and intermittent fever, several features were considered atypical for uncomplicated inflammatory orbital disease, including the rapid progression of a firm medial canthal mass, early proptosis, marked extraocular movement restriction, elevated intraocular pressure, and deterioration in visual function. Initial differential diagnoses included orbital cellulitis, paranasal sinus mucocele, lymphoma, and other malignant orbital tumours, prompting urgent cross-sectional imaging and biopsy.
Management
The patient was started on oral co-amoxiclav and listed for urgent orbital biopsy, performed on February 19, 2025, under local anaesthetic. A pre-septal approach was used to access the lesion within the medial orbit. The sample was sent for histological analysis. Immediate postoperative recovery was uneventful.
Histopathological findings revealed a “small round blue cell” tumour morphology. Immunohistochemical staining was consistent with RMS. The diagnosis was subsequently confirmed by the pathology team at Addenbrooke’s Hospital.
Following confirmation, the patient was referred urgently to the Oncology and Oculoplastics teams at Addenbrooke’s Hospital for staging and further management. At follow-up, sutures were removed, and the patient continued to experience diplopia and orbital discomfort.
Follow-Up
On review at Cambridge University Hospitals on March 4, 2025, there was a significant worsening of proptosis, chemosis, and orbital discomfort. Intraocular pressure in the left eye was 31 mmHg, and ocular motility remained markedly limited, with further reduction in visual acuity. This acute deterioration raised concern for either post-biopsy inflammatory oedema or rapid tumour progression, both of which carried a risk of optic nerve compression and permanent visual loss. These visual prognosis concerns prompted urgent multidisciplinary discussion with the oncology and oculoplastics teams, and interim management included intensive ocular lubrication and close monitoring.
Diagnosis
A multidisciplinary review was subsequently held at University College London Hospital (UCLH) on April 11, 2025, to discuss the case. The consensus confirmed the diagnosis of orbital RMS and recognised the potential for a therapy-related secondary malignancy given the patient’s oncological history. The team agreed on initiating proton beam radiotherapy as part of definitive management, aiming to deliver local tumour control while minimising radiation exposure to surrounding ocular and neural structures. A course of proton beam therapy was planned and scheduled at a tertiary radiotherapy centre, with close coordination between oncology, radiology, and ophthalmology services for ongoing review.
Discussion
Orbital RMS is uncommon in adults and usually manifests in children [1-3]. The diagnostic challenge in this case lies in the subtle and nonspecific early presentation. Upper eyelid swelling is a common finding in ophthalmic practice and is often attributed to benign causes such as infection or inflammation [4]. However, it was crucial in this case to identify red flag features, including the presence of minimal early proptosis and a palpable firm mass in the medial canthal region [5]. These subtle but significant findings prompted further investigation and were key to reaching the diagnosis.
This case underscores the need for clinicians to maintain a high index of suspicion for orbital RMS when evaluating young adults with rapidly progressive periocular swelling that may initially mimic infection, particularly in those with a history of childhood malignancy or bone marrow transplantation. The patient’s oncological background raised concern for a therapy-related secondary malignancy and lowered the threshold for aggressive investigation [3]. The tumour’s aggressiveness was reflected by the rapid progression of symptoms, including increasing proptosis and ocular discomfort over a short period.
The early stages may be deceptive, with preserved visual acuity and absence of an afferent pupillary defect masking the underlying severity. The ethmoidal origin with invasion into the orbit and paranasal sinuses reflects the embryonal subtype’s infiltrative behaviour. Biopsy remains essential for diagnosis, with histology demonstrating the characteristic small round blue cell morphology.
A prompt multidisciplinary approach involving ophthalmology, oncology, pathology, and radiotherapy teams was vital to guide management. Referral to a tertiary oncology centre allowed for appropriate staging and treatment planning, including proton beam radiotherapy, to achieve local control while minimising damage to surrounding ocular and neural structures.
Population-based analyses have demonstrated a lower incidence of RMS in adults compared with paediatric patients, and orbital involvement outside childhood is uncommon, with most descriptions arising from small orbital series and reviews [1-3]. Compared with these reports, our patient was notable for adult age, prior childhood malignancy treated with bone marrow transplantation, ethmoidal sinus origin with orbital invasion, and rapid progression mimicking inflammatory disease, ultimately requiring proton beam radiotherapy.
Conclusions
This case is notable for the occurrence of orbital RMS in a young adult with a history of childhood leukaemia treated with chemotherapy and bone marrow transplantation, raising concern for a therapy-related secondary malignancy. Orbital RMS in young adults is rare but should be considered in rapidly progressing orbital masses, particularly in patients with a history of childhood malignancy. While this report describes a single patient and therefore cannot support broad prognostic or therapeutic generalisations, it reinforces important clinical warning signs and diagnostic principles relevant to general practice. Early imaging, biopsy, and multidisciplinary referral are key to timely diagnosis and treatment. This case also highlights the importance of recognising post-biopsy symptom progression and the need for close postoperative monitoring.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rhabdomyosarcoma: review for the ophthalmologist Surv Ophthalmol Shields JA Shields CL 39574820031255932610.1016/s 0039-6257(02)00415-0 · doi ↗ · pubmed ↗
- 2Orbital rhabdomyosarcomas: a review Saudi J Ophthalmol Jurdy L Merks JH Pieters BR Mourits MP Kloos RJ Strackee SD Saeed P 1671752720132422798210.1016/j.sjopt.2013.06.004PMC 3770217 · doi ↗ · pubmed ↗
- 3Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients J Clin Oncol Sultan I Qaddoumi I Yaser S Rodriguez-Galindo C Ferrari A 339133972720091939857410.1200/JCO.2008.19.7483 · doi ↗ · pubmed ↗
- 4Common inflammatory and infectious conditions of the eyelid Dis Mon Gordon AA Danek DJ Phelps PO 1010426620203262268110.1016/j.disamonth.2020.101042 · doi ↗ · pubmed ↗
- 5Red flags in neuro-ophthalmology Community Eye Health Ogun O 6465292017 https://pmc.ncbi.nlm.nih.gov/articles/PMC 5365040/PMC 536504028381904 · pubmed ↗