Generation of Human Haematopoietic Model Cell Lines Revealed Distinct Replication Stress Tolerance Between Two Oncogenic KRAS Mutations, G12V and A146T
Mone Okuda, Ryotaro Kawasumi, Kayoko Tanaka, Kouji Hirota

TL;DR
Researchers created human cell lines to compare how two KRAS mutations affect replication stress and cancer cell survival, finding distinct responses that could guide targeted therapies.
Contribution
The study introduces isogenic haematopoietic cell lines to compare KRAS.A146T and KRAS.G12V mutations, revealing their distinct replication stress tolerance.
Findings
KRAS.A146T cells rely on PrimPol for survival under replication stress.
KRAS.G12V cells are hypersensitive to ATR-Chk1 inhibitors and nucleoside analogues.
Both mutations increase replication stress compared to wild-type KRAS.
Abstract
KRAS is one of the most frequently mutated genes in all human cancers, and its oncogenic mutation hotspots are glycine 12 (G12), glycine 13 (G13), glutamine 61 (Q61) and alanine 146 (A146). Among these hotspot mutations, A146 substitution mutations (A146X) occur relatively infrequently, except for haematopoietic and lymphoid cancers, suggesting that A146X causes intrinsically distinct KRAS signalling compared to other KRAS oncogenic alleles. However, due to the absence of model A146X cell lines derived from haematopoietic sources, the cellular mechanisms that cause the differences between KRAS.A146X and other common KRAS mutants, such as KRAS.G12X, remain largely unexplored. In this study, we developed a set of isogenic model haematopoietic cell lines expressing KRAS.A146T, KRAS.G12V and KRAS.G12G (non-mutated) from the endogenous locus by genetically modifying the human lymphoblastoid…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
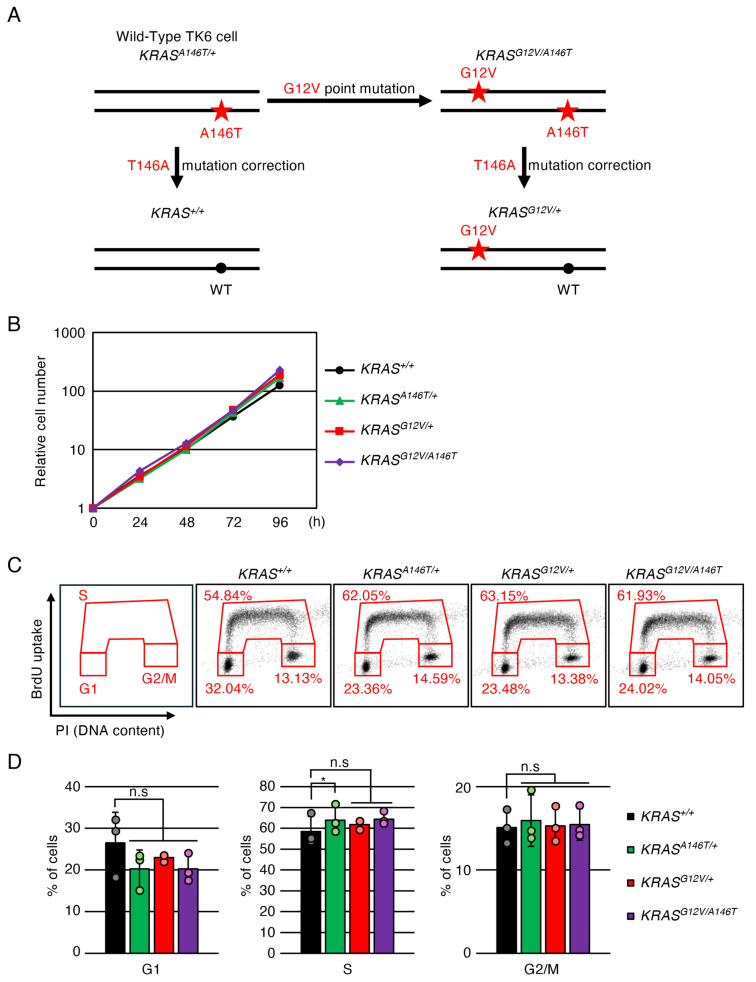 Figure 1
Figure 1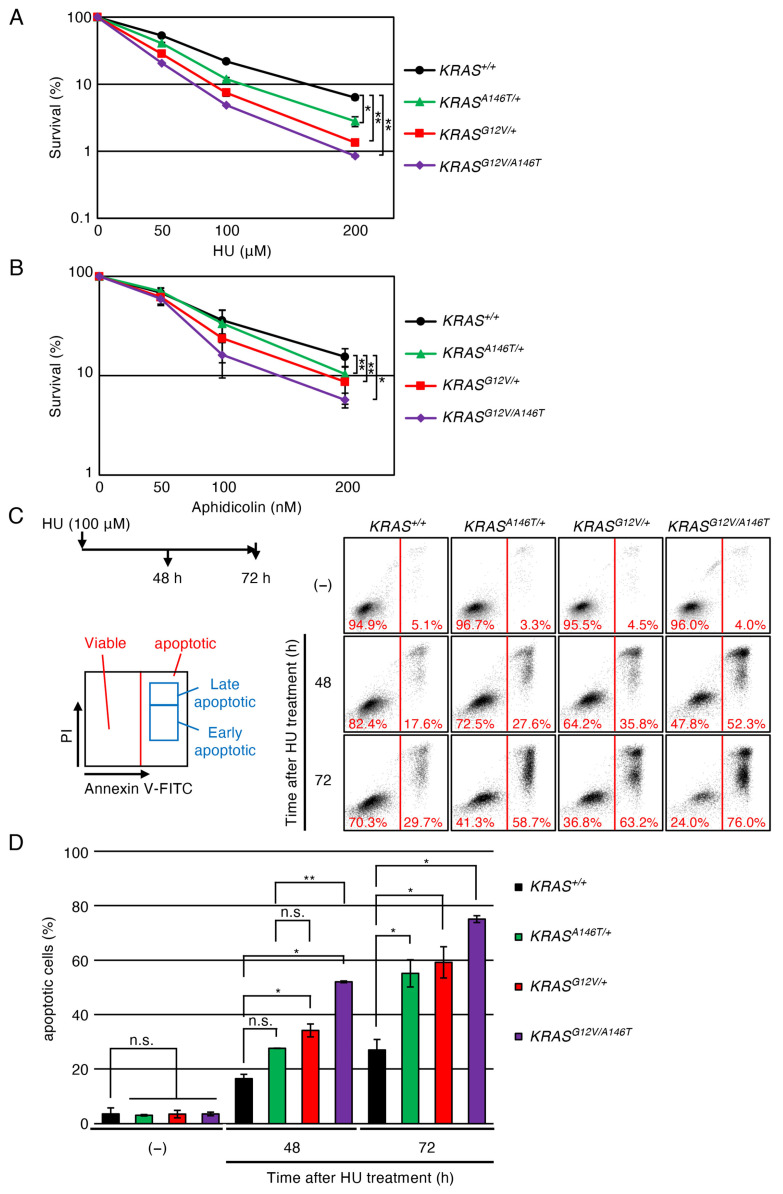 Figure 2
Figure 2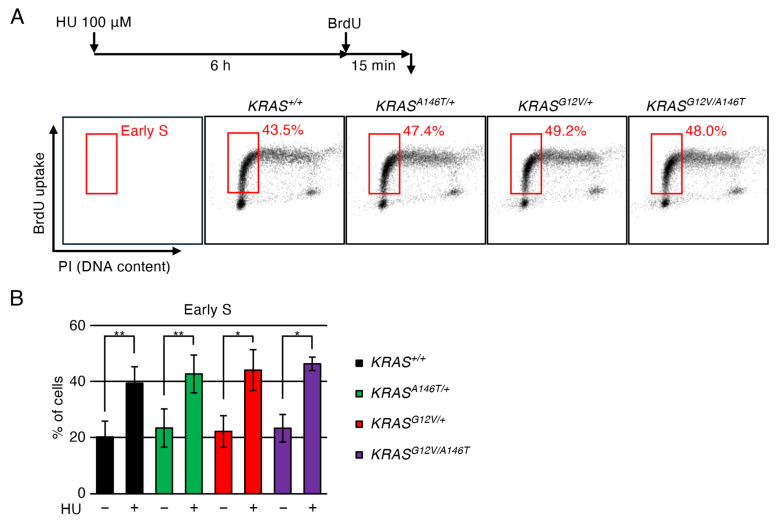 Figure 3
Figure 3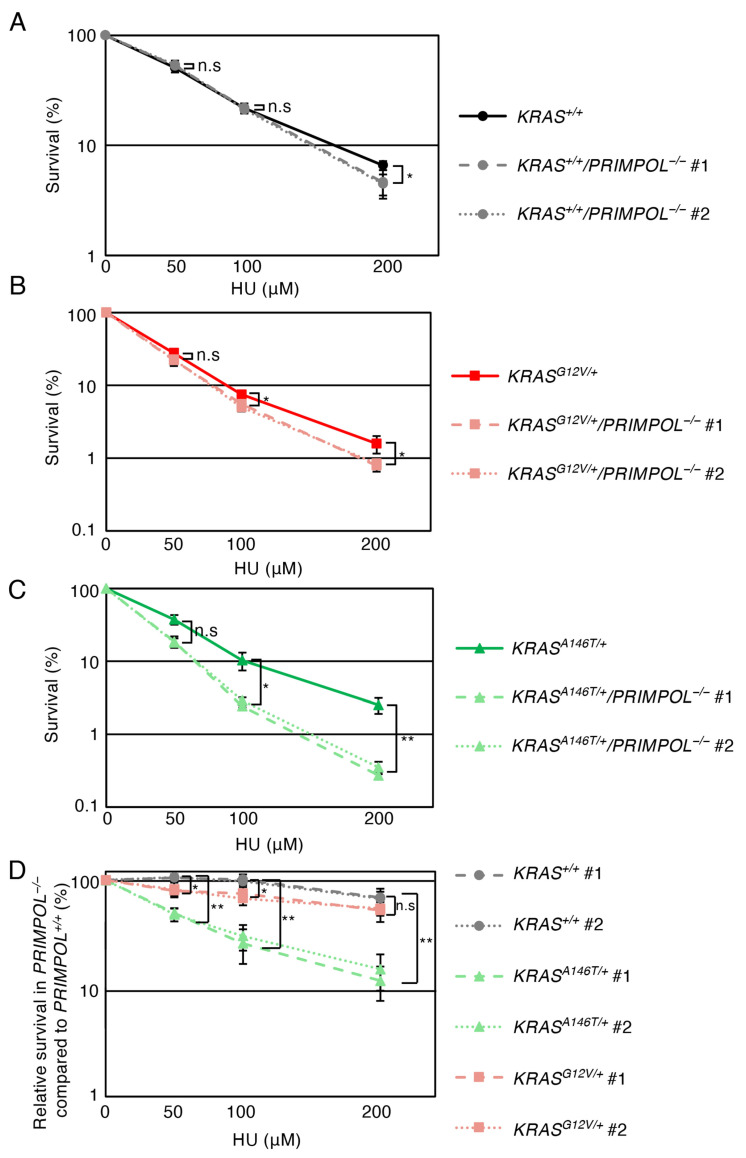 Figure 4
Figure 4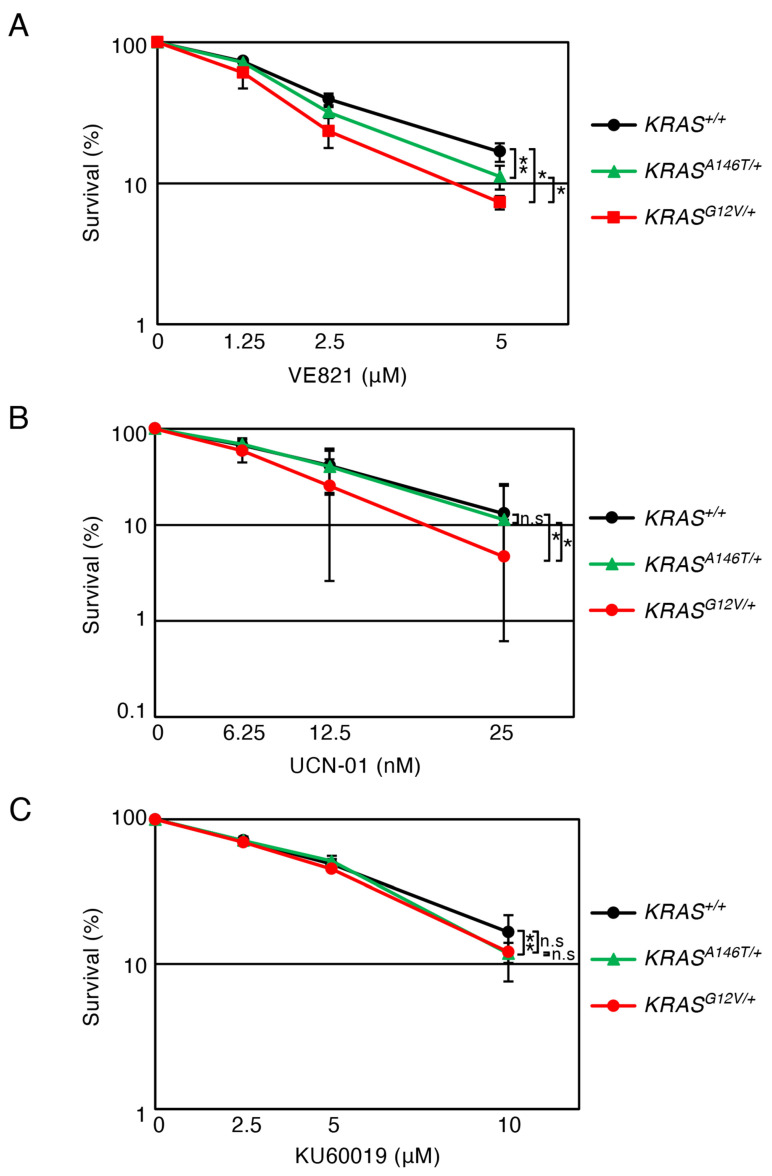 Figure 5
Figure 5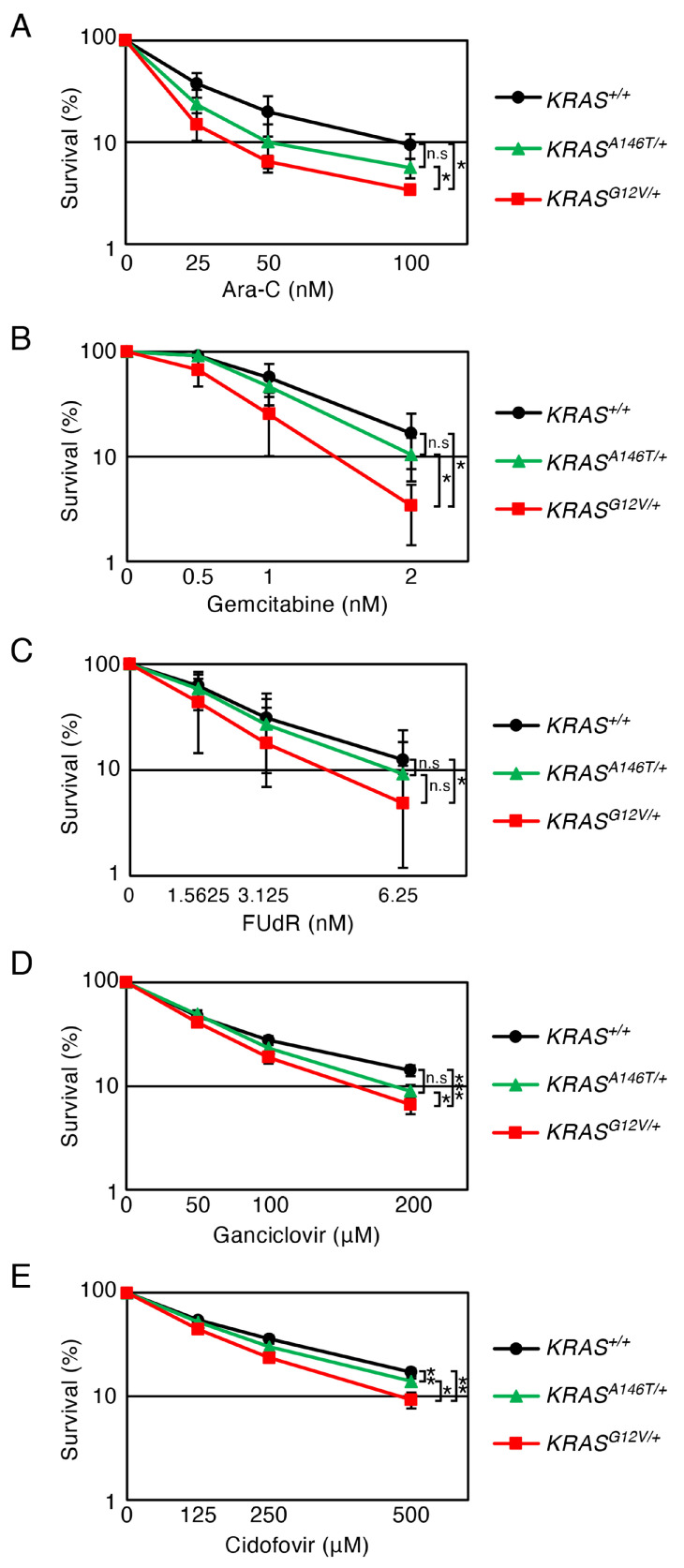 Figure 6
Figure 6- —JSPS KAKENHI
- —Tokyo Metropolitan Government Advanced Research
- —Takeda Science Foundation
- —Uehara Memorial Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Lymphoblastic Leukemia research · Acute Myeloid Leukemia Research · Protein Kinase Regulation and GTPase Signaling
1. Introduction
RAS proteins belong to the conserved RAS superfamily of small guanine nucleotide-binding proteins and play a pivotal role in signal transduction by serving as a molecular switch. They alternate between an active GTP-bound form and an inactive GDP-bound form, thereby regulating downstream signal pathways, including the mitogen-activated protein kinase (MAPK) pathway [1,2]. GTPase-activating protein (GAP) facilitates the hydrolysis of bound GTP by stimulating the intrinsic GTPase activity of RAS, shifting this switch into the inactive form, while guanine nucleotide-exchanging factor (GEF) induces the exchange of GDP to GTP, generating the activated RAS [3]. In humans, there are four RAS isoforms (HRAS, NRAS, KRAS4A and KRAS4B), encoded by three RAS genes, HRAS, NRAS and KRAS. Their activation is triggered by the growth factor-stimulated receptor tyrosine kinases (such as EGFR, FGFR and PDGFR) via GEF, transmitting signals to multiple downstream pathways to regulate cellular proliferation and differentiation [4,5,6,7].
The oncogenic potential of the RAS genes has been well established [8]. Approximately 25% of human RAS genes harbour mutations in all human cancers, with the KRAS gene being the most frequently mutated (approximately 18%) [9,10]. Oncogenic KRAS mutation hotspots are glycine 12 (G12), glycine 13 (G13), glutamine 61 (Q61) and alanine 146 (A146), and over 80% of all cancer-associated KRAS mutations occur at G12 (G12X), impairing the intrinsic and GAP-assisted GTP hydrolysis, leading to the accumulation of the GTP-bound active form [11,12]. Interestingly, the relative occurrence of these oncogenic KRAS alleles varies among different tissues and cancer types, indicating that different KRAS alleles generate distinct KRAS signalling. A prime example of such cases is the A146 substitution mutation (A146X). Whilst A146X is relatively infrequent, representing 1.1% of all KRAS-mutated cancers, its occurrence rate rises to 7.6% in haematopoietic/lymphoid cancers (COSMIC, v-102: https://www.cosmickb.org/, accessed on 1 January 2026). Interestingly, the most frequent A146X mutation, A146T, exhibits an increased intrinsic and GEF-induced nucleotide exchange capability, whereas it retains substantial GAP-assisted GTPase activity, clearly demonstrating distinct biochemical properties compared to the G12X variants [13]. However, the relationship between the biochemical property differences of the KRAS.A146X and KRAS.G12X variants and their cellular phenotypes is largely unexplored. Additionally, the reasons behind the tissue-specific allele prevalence patterns remain poorly understood. The absence of model KRAS oncogenic mutant cell lines derived from haematopoietic sources has hampered efforts to address these questions.
Earlier studies investigating the cellular consequences of oncogenic KRAS mutations have demonstrated various cancer-associated phenotypes, including augmented downstream signalling, increased replication stress [14,15,16] and enhanced cellular motility [17,18]. However, in most of these studies, phenotypic analyses relied primarily upon the overexpression of the mutated KRAS gene. As elegant KRAS mouse model studies have highlighted the importance of examining the effects of RAS mutations at the endogenous expression level [19,20,21,22], it would be important to revisit the oncogenic KRAS-induced cellular phenotypes using cultured cell systems where KRAS expression is maintained at physiological levels. However, only a limited number of human model cell lines endogenously expressing oncogenic KRAS variants have thus far been established [23]. Developing model cell lines of haematopoietic origin that express oncogenic KRAS alleles at physiological levels remains an important aspiration.
The lymphoblastoid TK6 cell line is a human haematopoietic-originated cell line, serving as a standard cell line for in vitro mammalian genotoxicity tests [24]. This cell line retains a stable near-diploid karyotype, except for a trisomic chromosome 13 [25]. The TK6 cells proliferate rapidly (with a doubling time of approximately 11–12 h) and maintain a consistent and reliable phenotype. Moreover, genetic modifications, including gene disruption and mutation using genome editing technologies such as CRISPR-Cas9 or TALEN [26,27], have been conducted in this cell line with relative ease. The information on the resultant Collection of Mutated Human cell lines from TK6 (COMHUT) was released by the TK6 consortium (https://www.nihs.go.jp/dgm/tk6.html, accessed on 1 January 2026). Since many researchers in the DNA replication, repair and recombination field have used this cell line, numerous mutants (deficient in DNA repair systems) have been created in this isogenic condition, and many of them can be obtained from COMHUT. We considered that adding the mutant cell lines harbouring oncogenic KRAS mutations to COMHUT could significantly advance our understanding of the effects of oncogenic KRAS mutations on DNA replication and DNA metabolism, as it would enable the utilization of known mutant cell lines.
We here report the generation of four isogenic human TK6 cell lines, where the endogenous KRAS alleles carry either KRAS^A146T^^/+^, KRAS^G12V^^/+^, KRAS^G12V^^/A146T^ or KRAS^+/+^. Strikingly, the KRAS^A146T^^/+^ and KRAS^G12V^^/+^ cell lines exhibited distinct replication phenotypes. The KRAS^A146T^^/+^ cells under stressed replication conditions are highly reliant on PrimPol, a polymerase/primase protein facilitating replication restart via repriming or translesion synthesis [28,29,30,31]. On the other hand, the KRAS ^G12V^^/+^ cells are hypersensitive to checkpoint inhibitors, VE821 (an ATR inhibitor) and UCN-01 (a Chk1 inhibitor), as well as to nucleoside analogues, Ara-C, gemcitabine and 5′-fluorodeoxyuridine, which are used in cancer chemotherapy and the treatment of viral infections [32,33,34,35]. These findings firstly highlight the profound effects of a single chromosomal copy of the oncogenic KRAS mutation on the replication process. Secondly, our observations provide strong evidence that KRAS.A146T and KRAS.G12V cause distinct cellular outcomes. The generated set of cell lines, aligned with COMHUT, serves as an invaluable tool for investigating the molecular mechanisms underlying allele-specific oncogenic KRAS signalling, which enhances our understanding of tissue-specific phenotypes and aids in the development of effective therapeutic reagents.
2. Materials and Methods
2.1. TK6 Cell Culture
TK6 cell line was obtained from the JCRB Cell Bank (accession number: JCRB 1435). TK6 cells were cultured as previously described [36]. Briefly, TK6 cells were cultured in Roswell Park Memorial Institute 1640 medium (Nacalai Tesque, Kyoto, Japan) supplemented with 10% heat-inactivated horse serum from Gibco and sodium pyruvate (1.8 mM), L-glutamine (2 mM), penicillin (100 U/mL) and streptomycin (100 μg/mL) from Nacalai Tesque.
2.2. Generation of Isogenic Cell Lines Carrying Oncogenic KRAS Mutation from TK6
For the knock-in of G12V mutation in the KRAS gene, KRAS gene was first disrupted with knock-in (KI) constructs prepared using primers 5′-GCGAATTGGGTACCGGGCCGAGATGGAGTCTTACTCCGTCACCCAATCT-3′ and 5′-CTGGGCTCGAGGGGGGGCCGCACAGAGAGTGAACATCATGGACCCTGAC-3′ for the left arm and 5′-TGGGAAGCTTGTCGACTTAACCAAAGTAAAGACCAGCAGCAGAATGATAAG-3′ and 5′-CACTAGTAGGCGCGCCTTAAATGTAATGTGTCAGTTCCCTCAGAGACTCA-3′ for the right arm. Underlined flanking sequences are used for seamless cloning. The second exon of the KRAS gene is in the left arm sequence. A point mutation (G12V) was introduced using the following primers: 5′-GTGGTAGTTGGAGCAGTGGGCGTAGGCAAGAGT-3′ and 5′-ACTCTTGCCTACGCCCACTGCTCCAACTACCAC-3′. The PCR-amplified left and right arms were inserted in marker-gene plasmids (DT-ApA/NEO^R^-based plasmids) digested with ApaI and AflII using the GeneArt Seamless Cloning & Gibson Assembly system (Thermo Fisher Scientific Inc., Waltham, MA, USA). The resultant KI plasmids express diphtheria toxin from outside of the homologous arms to suppress random integration events. The CRISPR expression vector for the CRISPR-Cas9 system was designed to recognize 5′-GTATTTCAGAGTTTCGTGAG-3′ (Figure S1). Parental TK6 cells were transfected with the above-mentioned targeting vectors (2 μg) and the expression vector (7 μg) for CRISPR using the NEON Transfection System (Thermo Fisher Scientific, Waltham, MO, USA) at 1400 V with a pulse width of 20 ms. The insertion of the maker gene was confirmed by PCR using the following primers: 5′-TTTATCAAATTTAGCGCTGTATTCACGCAG-3′ and 5′-ACTCTTGCCTACGCCCACTGCTCCAACTACCAC-3′. Then, the inserted maker gene was removed by expression of CRE recombinase. The introduction of a point mutation was confirmed by RT-PCR and following sequence analysis using the following primers: 5′-TCATTACGATACACGTCTGCAGTCAACTGG-3′ and 5′-CTGTATCGTCAAGGCACTCTTGCCTACGCC-3′.
For the removal of the A146T mutation in the KRAS gene, the KRAS gene was first disrupted with KI constructs prepared using primers 5′-GCGAATTGGGTACCGGGCCGTGCTGCTGCGAACATTGGTGTACATGTATC-3′ and 5′-CTGGGCTCGAGGGGGGGCCGGGTAAACTTGGATAATAGAGCTGAAATTTGG-3′ for the left arm and 5′-TGGGAAGCTTGTCGACTTAACTAGGTATTTGATCTTTTGAGAGAGATACAAGG-3′ and 5′-CACTAGTAGGCGCGCCTTAATGCTTAGTGATGTTATAGCCATCCTAACAC-3′ for the right arm. The 3rd exon of the KRAS gene is in the left arm sequence. A point mutation (T146A) was introduced using the following primers: 5′-ATTGAAACATCAGCAAAGACAAGAC-3′ and 5′-GTCTTGTCTTTGCTGATGTTTCAAT-3′. The PCR-amplified left and right arms were inserted in marker-gene plasmids (DT-ApA/NEO^R^-based plasmids) digested with ApaI and AflII using the GeneArt Seamless Cloning & Gibson Assembly system (Thermo Fisher Scientific, Waltham, MO, USA). The CRISPR expression vector for the CRISPR-Cas9 system was designed to recognize 5′-CACCAGCTAATGGTGTTCGGAACC-3′ (Figure S2). Parental TK6 cells were transfected as described above. The insertion of the maker gene was confirmed by PCR using the following primers: 5′-TTTATCAAATTTAGCGCTGTATTCACGCAG-3′ and 5′-CATGTACCACATAATGACATTTTGGTCCAC-3′. Then, the inserted maker gene was removed as described above.
2.3. Measurement of Cellular Sensitivity to Drugs
To measure cellular sensitivity to replication inhibitors, nucleoside analogues and low-molecular-weight inhibitors, a liquid-culture cell-survival assay was employed as previously described [37]. Briefly, TK6 cells were diluted in medium (10^4^ cells/mL) and dispensed into a 24-well plate (1 mL) to which the above drugs were added and mixed before culturing for 72 h. The incubated cells (100 μL) were then transferred to 96-well plates, and the amount of ATP was measured using the CellTiter-Glo Cell Viability Assay (Promega, Madison, WI, USA), according to the manufacturer’s instructions. Luminescence was measured using a Fluoroskan Ascent FL Microplate Fluorometer and Luminometer (Thermo Fisher Scientific Inc., Waltham, MA, USA).
2.4. Cell-Cycle Analysis
Cell-cycle distribution was assessed using flow cytometry as previously described [38]. Briefly, cells were pulse-labelled with 10 μM BrdU for 15 min and fixed in 70% ethanol. After permeabilization and denaturation by treating cells with 0.5% Triton X-100 and 2 N HCl, cells were subjected to immunostaining using anti-BrdU antibody (cat. no. 347580; Becton, Dickinson and Company, Franklin Lakes, NJ, USA, 1:100 dilution) and secondary antibody conjugated with Alexa488 (cat. no. 115-545-003; Jackson ImmunoResearch West Grove, PA, USA, 1:50 dilution). Finally, cells were stained with propidium iodide and analysed using a BD Accuri™ C6 flow cytometer (Becton Dickinson, NJ, USA).
2.5. Measurement of Apoptotic Cell Fraction Using Flowcytometry
Flow cytometric analysis of apoptosis was performed using an Annexin V-FITC apoptosis detection kit (15342-54; Nacalai Tesque, Kyoto, Japan) according to the manufacturer’s instructions. Cells (1 × 10^5^) were harvested at 0, 48 and 72 h after HU treatment (0.1 mM). The cells were washed twice with PBS and resuspended in 100 μL of Annexin V binding buffer. Then, 5 μL each of Annexin V-FITC solution and propidium iodide solution were added, followed by incubation in the dark for 15 min. Subsequently, 400 μL of Annexin V binding buffer was added, and the samples were analysed by flowcytometry (BD Accuri C6 Plus; Becton, Dickinson and Company, Franklin Lakes, NJ, USA). Flowcytometric analysis was performed after gating to exclude debris and events smaller than intact cells. Despite the small sample size, the data maintained normality, allowing us to perform statistical analysis using t-tests on two independent datasets [39].
2.6. Chromosomal Aberration Analysis
Mitotic chromosome spreads were prepared and analysed as described previously [37]. TK6 cells were treated with 0.1 μg/mL colcemid (15212012; Thermo Fisher Scientific, Waltham, MA, USA) for the last 3 h.
2.7. Measurement of γH2AX-Positive Cell Fraction Using Flowcytometry
Cells (a total of 5 × 10^5^) were harvested. The cells were washed once with PBS, resuspended in 500 μL of cold 70% ethanol and fixed at 4 °C for at least 1 h. After fixation, the cells were washed once with 500 μL of 1% BSA/PBS, treated with 500 μL of 0.5% Triton/PBS and incubated at room temperature for 10 min. They were then washed once with 500 μL of 1% BSA/PBS, resuspended in 30 μL of 1% BSA/PBS containing 0.6 μL of anti-γH2AX (mouse) antibody (05-636; Millipore, Burlington, MA, USA) and incubated at room temperature for 60 min with gentle tapping every 15 min. The cells were washed twice with 500 μL of 1% BSA/PBS, resuspended in 30 μL of 1% BSA/PBS containing 0.3 μL of Alexa488 conjugated anti-mouse antibody (A11001; Thermo Fisher Scientific, Waltham, MA, USA) and incubated again at room temperature for 60 min with gentle tapping every 15 min. After two additional washes with 500 μL of 1% BSA/PBS, 500 μL of 1% BSA/PBS containing 50 μg/mL RNase A (313-01461; NIPPON GENE, Tokyo, Japan) and 5 μg/mL propidium iodide (PI (169-26281; FUJIFILM, Osaka, Japan)) was added. The cells were then incubated at 37 °C for 30 min, passed through a cell strainer and analysed by flowcytometry (BD Accuri C6 Plus; Becton, Dickinson and Company, Franklin Lakes, NJ, USA). Flowcytometric analysis was performed after gating to include viable cells.
3. Results
3.1. Establishment of Isogenic KRASG12V/+, KRASA146T/+, KRASG12V/A146T and KRAS+/+ Cells from TK6
The presence of the A146T mutation in the KRAS gene in TK6 had been anticipated, since this KRAS mutation is detected in WIL2-NS cells (https://depmap.org/portal/cell_line/ACH-002316?tab=overview, accessed on 1 January 2026).), which are derived from WI-L2 (a cell line derived from the spleen of a 5-year-old boy suffering from hereditary spherocytosis [40]), the original cell line for TK6 [41]. Indeed, our initial sequence analysis of KRAS cDNA revealed that parental TK6 cells harbour a heterozygous A146T mutation in the KRAS gene. Given its reported oncogenic potential [13,42], we referred to the original TK6 cells as KRAS^A146T^^/+^ cells in this study. To introduce an additional oncogenic mutation, we inserted a point mutation (G12V) into the non-mutated KRAS allele of the KRAS^A146T^^/+^ cells, thereby establishing KRAS^G12V/A146T^ cells (Figure S1A–D). Expression of the KRAS.G12V transcript in this cell line was confirmed by RT-PCR, followed by sequencing analysis (Figure S1E). Subsequently, we corrected the A146T mutation using the KRAS.T146A knock-in construct in both KRAS^A146T^^/+^ and KRAS^G12V/A146T^ cells, resulting in the generation of KRAS^+/+^ and KRAS^G12V^^/+^ cells, respectively (Figure 1A and Figure S2). We compared the proliferation speed among KRAS^+/+^, KRAS^G12V^^/+^, KRAS^A146T^^/+^ and KRAS^G12V/A146T^ cells, and found that growth speed was comparable in these cell lines (Figure 1B). We further analysed the effects of oncogenic KRAS expression on the cell cycle. In cells expressing the oncogenic KRAS genes, a slight decrease in the G1-phase population and an increase in the S-phase population were observed. However, these changes were not statistically significant, suggesting that the overall cell-cycle profile of TK6 cells was not substantially affected by oncogenic KRAS expression (Figure 1C,D).
3.2. Increased Replication Stress Vulnerability in KRASG12V/+, KRASA146T/+ and KRASG12V/A146T TK6 Cells
To assess the impact of the oncogenic KRAS mutations on the replication process, we analysed the established isogenic TK6 cell lines for their cellular sensitivity to hydroxyurea (HU), a replication stress-inducing agent that transiently stalls replication forks by depleting deoxyribonucleotide pools [43]. Both KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells exhibited increased sensitivity to HU compared to KRAS^+/+^ cells (Figure 2A), suggesting that these mutations compromise replication robustness. This observation aligns well with previous reports demonstrating elevated replication stress in cells expressing oncogenic KRAS [17,23,44]. Moreover, KRAS^G12V/A146T^ cells exhibited additively augmented HU sensitivity compared to KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells, indicating a potential correlation between oncogenic KRAS gene dosage and replication stress vulnerability. These oncogenic KRAS-expressing TK6 cells also showed hypersensitivity to aphidicolin, an inhibitor for replicative polymerases, confirming that the endogenously expressed oncogenic KRAS mutations likely exacerbate replication stress [45] (Figure 2B). To investigate whether the augmented HU sensitivity in oncogenic KRAS-expressing cells is attributable to enhanced apoptosis induced by HU, we analysed populations of the early and late apoptotic cells using Annexin V staining. We found that the proportion of early and late apoptotic cells was increased in KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells compared to KRAS^+/+^ cells at 72 h after HU treatment. Furthermore, KRAS^G12V/A146T^ cells showed an additive increase in apoptosis compared to KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells (Figure 2C,D). Taken together, these results indicate that these oncogenic KRAS mutations compromise replication robustness.
Next, we investigated whether the increased cell death during replication stress in oncogenic KRAS-expressing cells was attributable to checkpoint deficiency. To this end, we analysed the effects of HU treatment on the cell cycle. We found that both KRAS^+/+^ and oncogenic KRAS-expressing cells similarly exhibited accumulation in early S-phase following HU treatment (Figure 3). These results indicate that the replication stress checkpoint is not impaired in oncogenic KRAS-expressing cells. We further investigated the activation status of replication checkpoints. To this end, we monitored the phosphorylation status of Chk1 upon HU treatment. Consistent with the active cell-cycle arrest in oncogenic KRAS-expressing cells upon HU, these cells exhibited similar levels of phosphorylated Chk1 (Chk1-p) after HU treatment compared to KRAS^+/+^ cells (Figure S3). These data indicate that the ATR-Chk1 checkpoint pathway is not compromised in these KRAS-expressing cells. Importantly, under unperturbed conditions, the levels of Chk1-p in these KRAS-expressing cells were also indistinguishable from those in KRAS^+/+^ cells. These data suggest that endogenous expression of oncogenic KRAS perturbs DNA replication, but not to an extent sufficient to induce checkpoint activation (Figure S3). Similarly, in these KRAS-expressing cells, no increase in Histone H2AX phosphorylation (γH2AX, a marker of DNA replication stress [46]) or in the number of chromosome aberrations was detected (Figures S4 and S5).
3.3. PrimPol Is Required for the Proliferation of KRASA146T/+ Cells in HU-Stressed Replication Condition
Having established that replication stress is exacerbated in the oncogenic KRAS-expressing TK6, we next investigated the role of PrimPol, a DNA polymerase/primase involved in alleviating replication fork stress by facilitating replication fork restart [28,29,30,31], in sustaining cell viability when the cells are exposed to the HU-induced replication stress. A connection between PrimPol and the oncogenic KRAS signalling has been indicated by a recent study that highlighted the role of PrimPol in maintaining continuous DNA replication in cells where the replication process is disturbed by the ectopic expression of the oncogenic KRAS.G12V variant [17]. We disrupted the PRIMPOL gene (PRIMPOL^−/−^) in KRAS^+/+^, KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells and assessed the effect of the loss of PrimPol on HU sensitivity in each gene status. The loss of PrimPol in KRAS^+/+^ or KRAS^G12V^^/+^ cells had a limited impact on the HU sensitivity (Figure 4A,B). In contrast, KRAS^A146T^^/+^ cells exhibited significantly higher HU sensitivity when the PRIMPOL gene was disrupted (Figure 4C). These results show that the requirement of PrimPol for alleviating replication stresses is greater in KRAS^A146T^^/+^ than that in KRAS^+/+^ and KRAS^G12V^^/+^ cells (Figure 4D), demonstrating distinct replication phenotypes between the KRAS^A146T^^/+^ and KRAS^G12V^^/+^ cells.
3.4. Effects of Checkpoint Inhibitors on Oncogenic KRAS-Expressing Cells
Given the critical role of checkpoint pathways in maintaining replication integrity [47,48], we next examined the effect of checkpoint inhibition on the cellular survival of the KRAS^G12V^^/+^, KRAS^A146T^^/+^ and KRAS^+/+^ cells. We tested VE821 (an ATR inhibitor [49]), UCN-01 (a Chk1 inhibitor [50]) and KU60019 (an ATM inhibitor [51]). Both KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells exhibited significantly higher sensitivity to VE821 than KRAS^+/+^ cells. Interestingly, the KRAS.G12V mutation exhibited a greater impact (over 55% reduction in survival rate) than the KRAS.A146T mutation (35% reduction in survival rate). To UCN-01, KRAS^G12V^^/+^ cells, but not KRAS^A146T^^/+^, exhibited a significantly increased sensitivity, decreasing the survival rate by more than 60% (Figure 5A,B). These data suggest that KRAS.G12V causes replication stress, making cells rely on the ATR-Chk1 checkpoint axis, whereas KRAS.A146T exerts a limited effect on the ATR-Chk1 checkpoint. Responses to the ATM inhibitor KU60019 also highlighted the difference between KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells; the KRAS^G12V^^/+^ cells exhibited significantly increased sensitivity to KU60019 compared to KRAS^+/+^ cells, whereas the KRAS^A146T^^/+^ cells responded similarly to the KRAS^+/+^ cells. Interestingly, the KU60019 treatment caused only less than a 30% reduction in the KRAS.G12V cells’ survival rate, suggesting that ATM inhibition has limited impacts (Figure 5C). Taken together, these results indicate that replication stress induced by KRAS.G12V is likely distinct from that induced by KRAS.A146T. Furthermore, KRAS oncogenic mutations are unlikely to severely challenge the ATM checkpoint.
3.5. Effects of the Chain-Terminating Nucleoside Analogues on Oncogenic KRAS-Expressing Cells
To identify therapeutic agents selectively targeting cells with augmented replication stress due to oncogenic KRAS expression, we evaluated the sensitivity of KRAS-mutant cell lines to chain-terminating nucleoside analogues (CTNAs). These drugs are incorporated into DNA during DNA synthesis, leading to premature chain termination, and are commonly used in cancer treatments and antiviral therapies [52,53,54]. In this experiment, we tested five CTNAs: Ara-C (a cytidine analogue, aka cytarabine, used in leukaemia treatment [33,34]), gemcitabine (a cytidine analogue, used in the treatment of various cancers [55,56]), 5-fluorodeoxyuridine (a uridine analogue, aka FUdR, used in the treatment of various cancers [35]), ganciclovir (a guanosine analogue, used in the treatment of viral infection [57,58]) and cidofovir (a cytidine analogue, used in the treatment of viral infection [31]). Against all tested CTNAs, KRAS^G12V^^/+^ cells exhibited significantly higher sensitivity than KRAS^+/+^ cells (Figure 6A–E). Meanwhile, KRAS^A146T^^/+^ cells responded to these CTNAs in a comparable manner to the KRAS^+/+^ cells, except for cidofovir, which induced a mild reduction in the survival rate (less than 20%) in the KRAS.A146T cells (Figure 6A–E). Collectively, these results demonstrate that a wide range of CTNAs tested in this study effectively reduce cell viability in cells expressing oncogenic KRAS.G12V, whereas they show limited efficacy against KRAS.A146T cells.
4. Discussion
In this study, we established isogenic TK6-derived cell lines carrying oncogenic KRAS mutations (KRAS^+/+^, KRAS^G12V^^/+^, KRAS^A146T^^/+^ and KRAS^G12V/A164T^ cells) (Figure 1). To our knowledge, these are the first isogenic set of haematopoietic-originated cell lines harbouring KRAS oncogenic variants, including A146T. We found that one chromosomal copy of KRAS.G12V or KRAS.A146T renders cells hypersensitive to replication stress-inducing agents, such as HU and aphidicolin, indicating that these oncogenic KRAS mutations induce replication fragility in human TK6 cells (Figure 2 and Figure 3). We further found that PrimPol, a primase/polymerase enzyme implicated in the restart of stalled replication [28,29], plays a pivotal role in alleviating replication stress induced in KRAS^A146T^^/+^ cells (Figure 4). Further, we found that the inhibition of the ATR-Chk1 pathway, but not the ATM pathway, reduced the viability of the KRAS^G12V^^/+^ cells (Figure 5). Finally, we found that a range of CTNAs induce cytotoxicity in cells expressing oncogenic KRAS.G12V (Figure 6).
Our observation that both KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells exhibit augmented sensitivity to HU compared to KRAS^+/+^ cells (Figure 2) is consistent with our previous study, where the human hTERT RPE-1 cells harbouring either KRAS^G12V^^/+^, KRAS^G12C^^/+^ or KRAS^G12D^^/+^ were sensitive to HU [23]. These results support the notion that one chromosomal copy of oncogenic KRAS mutations is sufficient to induce replication stress vulnerability. We also observed that the cells harbouring KRAS^G12V/A146T^ exhibited an increased HU/aphidicolin sensitivity compared to the KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells (Figure 2). This suggests that a higher copy number of oncogenic KRAS is linked to increased replication stress. Previous studies have shown that the ectopic overexpression of oncogenic RAS leads to hyper-replication, unscheduled activation of replication origins, unstable replication machinery and DNA damage on the template strands [14,44,59,60]. It is of great interest whether the replication phenotypes observed in this study, caused by the endogenous KRAS mutations, represent a milder version of the “RAS overexpression” phenotypes or whether they are fundamentally different from the “overexpression” cases.
Although the expression of oncogenic KRAS mutations tested here in endogenous levels compromised replication robustness (Figure 2), no detectable increase in Chk1 or H2AX phosphorylation levels was observed in unperturbed conditions (Figure 4 and Figure S3). Moreover, chromosome aberrations were not increased in these oncogenic KRAS-expressing cells either (Figure S5). These results are reminiscent of a previous study showing that low replication stress does not activate the ATR-Chk1 signalling axis, but still perturbs fork progression, leading to chromosome instability, notably at common fragile sites [61]. These findings suggest a new theory for oncogenesis—low-level replication stress that does not activate checkpoints promotes gradual genomic instability, leading to transformation—in addition to the previously appreciated model where inactivation of DNA repair mechanisms and checkpoints promotes oncogene-driven transformation [62].
PrimPol-mediated replication restart mechanisms have been proposed to play a key role in fork recovery [29,63,64]. Our data revealed a synergistic interaction between the oncogenic KRAS mutations and PrimPol deficiency under HU-induced replication stress, with this effect being particularly pronounced in the KRAS.A146T case (Figure 4D). This suggests that targeting PrimPol in combination with HU treatment might be a strategy against KRAS.A164T-derived haematopoietic and lymphoid cancers. Meanwhile, it has to be noted that the synergistic effect of HU and the loss of PrimPol did not cause a profound effect on the KRAS.G12V-mutant cell line, underlining the importance of considering the KRAS allele-specific effect when developing treatment strategies. It remains unclear why KRAS.A146T and KRAS.G12V proteins affect the replication process differently. Further studies are required to understand the mechanism of their distinct impacts on replication.
Oncogenic mutations at A146 of KRAS are observed with notable frequency exclusively within haematopoietic and lymphoid tissues (COSMIC, v102). Previous studies comparing the effect of KRAS.G12D—a common mutant type—and KRAS.A146T—a tissue-specific variant—have shown that the effect of KRAS.A146T resembles that of KRAS.G12D, albeit in a milder form [13,42]. In contrast, our current study, employing the model oncogenic KRAS-mutant cell lines derived from the human haematopoietic lymphoblastoid TK6 cell line, revealed that KRAS.A146T mutation imposes a significantly greater dependency upon PrimPol for cell survival under conditions of replication stress than does KRAS.G12V mutation. One interpretation of the greater dependency on PrimPol is an elevated frequency of repriming events. We propose that such repriming events in KRAS.A146T-mutated cells of haematopoietic origin might generate extensive single-stranded gaps in the genome, thereby promoting genome instability, which in turn may underpin the biological selection favouring specific KRAS mutations. The mechanism by which the KRAS.A146T mutation, which promotes GDP-GTP exchange, activates PrimPol remains an important question to be elucidated in the future. Similar to A146X, the substitution of glycine 13 into aspartic acid (G13D) is frequently observed in haematopoietic and lymphoid tissues. Intriguingly, the KRAS.G13D protein also displays an augmented rate of GDP-GTP exchange [12,65], as KRAS.A146T does, suggesting that its oncogenic potency is likely governed by a mechanism similar to that of KRAS.A146T [13]. Further investigation of KRAS.G13D in TK6 cells might give a clue to understand the mechanistic basis for the pronounced dependency on PrimPol in cells of haematopoietic origin, where KRAS hyperactivation occurs through accelerated nucleotide exchange.
We identified agents that reduce the cellular survival of the oncogenic KRAS-expressing cells. We found that an ATR inhibitor (VE821) and a Chk1 inhibitor (UCN-01) efficiently reduced cellular survival in KRAS^G12V^^/+^ (Figure 5). Moreover, several CTNAs preferentially reduced cellular viability of KRAS^G12V^^/+^ cells compared to KRAS^+/+^ cells (Figure 6). Furthermore, since the loss of PrimPol critically reduced the survival rate of KRAS^A146T^^/+^ cells under stressed replication condition (Figure 4), the inhibitors of PrimPol are expected to reduce cellular survival in KRAS^A146T^^/+^ cells. Given the widespread prevalence of KRAS mutations in various cancers and their well-documented association with adverse clinical outcomes and therapeutic resistance [66,67], our findings have important translational implications. The identification of synthetic vulnerabilities—such as PrimPol dependency or hypersensitivity to checkpoint inhibitors and chain-terminating nucleoside analogues (CTNAs)—in oncogenic KRAS-expressing cells provides valuable avenues for the development of novel therapeutic strategies targeting replication stress of KRAS mutated cancers. Notably, the distinct responses observed between KRAS^G12V^^/+^ and KRAS^A146T^^/+^ cells underscore the necessity of accounting for mutation-specific effects when designing precision treatments.
5. Conclusions
The isogenic TK6 oncogenic KRAS model established in this study offers a robust platform for dissecting molecular mechanisms underlying replication stress and for conducting high-throughput compound screens aimed at selectively eradicating KRAS-mutant cells, thereby advancing precision oncology initiatives. Due to its rapid proliferation property and ease of culturing (suspension cells), TK6 can be one of the ideal screening platforms. Furthermore, by utilising the KRAS mutation KI systems and guiding the RNAs developed herein to generate double-mutant derivatives of cell lines listed in COMHUT (TK6 Consortium: https://www.nihs.go.jp/dgm/tk6.html, accessed on 1 January 2026), it may be possible to further elucidate the molecular basis of replication vulnerability instigated by oncogenic KRAS.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alharbi S. Merkle S. Hammill A.M. Waters A.M. Le Cras T.D. RAS Pathway Mutations and Therapeutics in Vascular Anomalies Pediatr. Blood Cancer 202572 e 3160510.1002/pbc.3160539984187 PMC 12916055 · doi ↗ · pubmed ↗
- 2Serna-Blasco R. Sanz-Álvarez M. AguileraÓ. García-Foncillas J. Targeting the RAS-dependent chemoresistance: The Warburg connection Semin. Cancer Biol.201954809010.1016/j.semcancer.2018.01.01629432815 · doi ↗ · pubmed ↗
- 3Hennig A. Markwart R. Esparza-Franco M.A. Ladds G. Rubio I. Ras activation revisited: Role of GEF and GAP systems Biol. Chem.201539683184810.1515/hsz-2014-025725781681 · doi ↗ · pubmed ↗
- 4Gimple R.C. Wang X. RAS: Striking at the Core of the Oncogenic Circuitry Front. Oncol.2019996510.3389/fonc.2019.0096531681559 PMC 6798062 · doi ↗ · pubmed ↗
- 5Muñoz-Maldonado C. Zimmer Y. MedováM. A Comparative Analysis of Individual RAS Mutations in Cancer Biology Front. Oncol.20199108810.3389/fonc.2019.0108831681616 PMC 6813200 · doi ↗ · pubmed ↗
- 6Rebollo A. Martínez A.C. Ras proteins: Recent advances and new functions Blood 1999942971298010.1182/blood.V 94.9.297110556179 · doi ↗ · pubmed ↗
- 7Vojtek A.B. Der C.J. Increasing complexity of the Ras signaling pathway J. Biol. Chem.1998273199251992810.1074/jbc.273.32.199259685325 · doi ↗ · pubmed ↗
- 8Downward J. Targeting RAS signalling pathways in cancer therapy Nat. Rev. Cancer 20033112210.1038/nrc 96912509763 · doi ↗ · pubmed ↗