Design, synthesis and antiproliferative activity of oxadiazole derivatives as potent glycogen synthase kinase-3/histone deacetylase 6 dual inhibitors
Changchun Ye, Zilu Chen, Jiantao Jiang, Jianzhong Li, Ranran Kong, Shiyuan Liu, Xin Chen, Zhengshui Xu

TL;DR
Scientists designed a compound that inhibits two enzymes linked to cancer, showing strong activity in tests.
Contribution
A new oxadiazole compound (15i) was developed as a dual inhibitor of GSK3 and HDAC6 with high potency.
Findings
Compound 15i has IC50 values of 5.50 nM for HDAC6 and 69 nM and 88 nM for GSK3α and GSK3β, respectively.
15i showed potent cytotoxicity against AGS cancer cells with submicromolar IC50 values.
Molecular docking simulations confirmed that 15i fits well into the active sites of HDAC6 and GSK3β.
Abstract
A series of oxadiazole-based dual inhibitors targeting GSK3 and HDAC6 were rationally designed by integrating key pharmacophores into a single molecule. Among these derivatives, 4-(((5-(benzo[d][1, 3]dioxol-5-yl)-1,3,4-oxadiazol-2-yl)thio)methyl)-N-hydroxybenzamide (15i) was identified as the most potent compound with IC50 of 5.50, 69 nM and 88 nM against HDAC6, GSK3α and GSK3β, respectively. 15i also exhibited potent cytotoxicity against the AGS cancer cell line, with IC50 values in the submicromolar range. Molecular docking simulation confirmed that 15i fitted well into the active sites of both HDAC6 and GSK3β. These findings establish compound 15i as a promising candidate for further evaluation.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
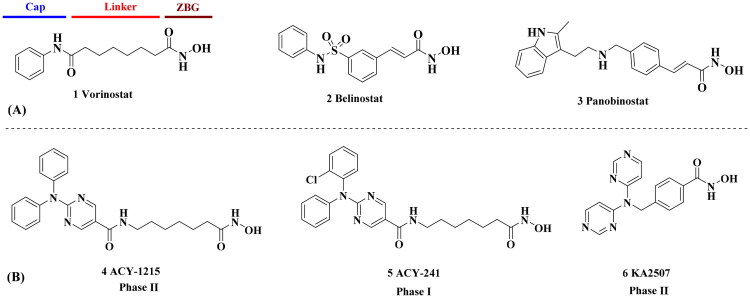 Figure 1
Figure 1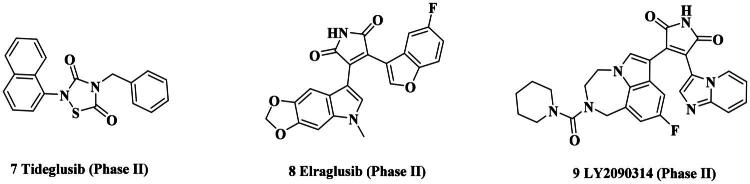 Figure 2
Figure 2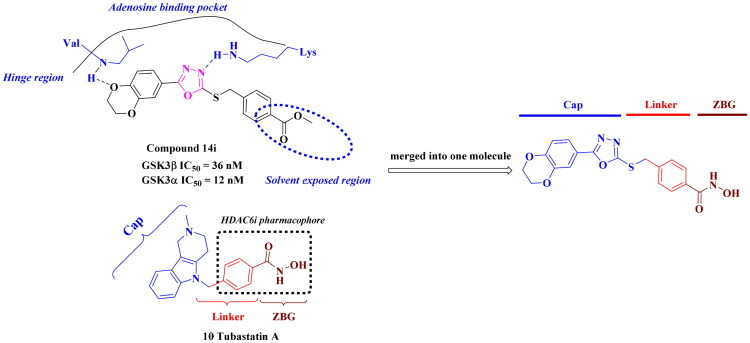 Figure 3
Figure 3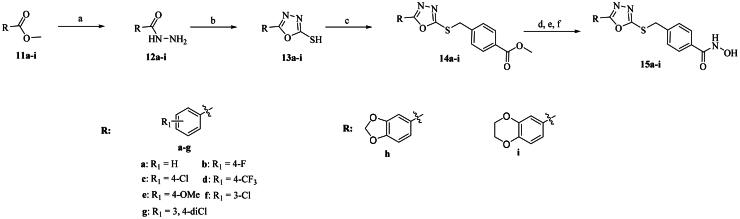 Scheme 1
Scheme 1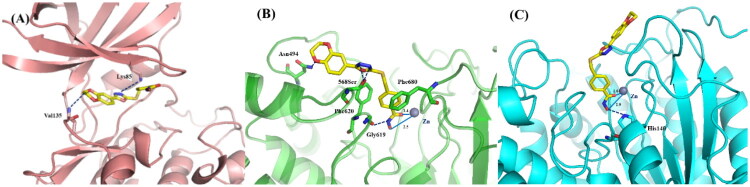 Figure 4
Figure 4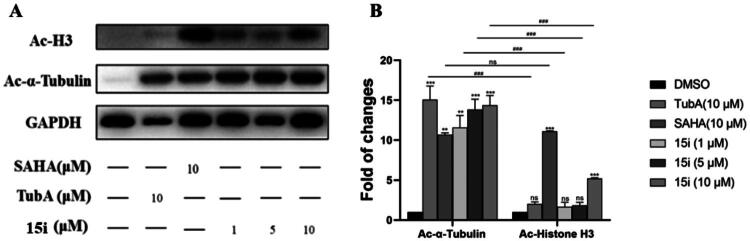 Figure 5
Figure 5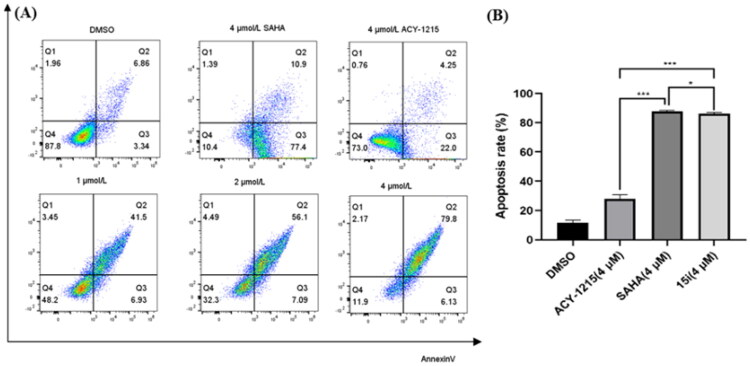 Figure 6
Figure 6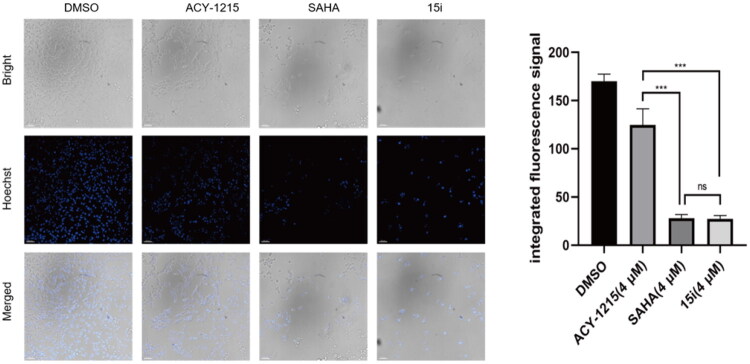 Figure 7
Figure 7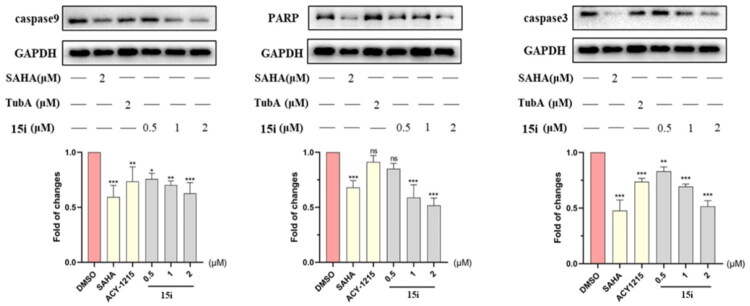 Figure 8
Figure 8|
| |||
|---|---|---|---|
| Compound | Het | HDAC6 | GSK3 |
|
|
| 12.3 ± 0.76 | 430 ± 26 |
|
|
| 8.60 ± 0.44 | 340 ± 30 |
|
|
| 9.00 ± 0.50 | 380 ± 19 |
|
|
| 7.10 ± 0.33 | 225 ± 10 |
|
|
| 6.00 ± 0.20 | 176 ± 9.7 |
|
|
| 11.0 ± 0.85 | 268 ± 21 |
|
|
| 8.45 ± 0.42 | 130 ± 8.5 |
|
|
| 7.06 ± 0.55 | 95 ± 3.0 |
|
|
| 5.50 ± 0.23 | 88 ± 5.4 |
|
| / | 5.13 ± 0.28 | / |
|
| / | 14.3 ± 0.41 | / |
|
| / | 7.10 ± 0.15 | / |
|
| / | / | 36 ± 1.1 |
|
| / | / | 710 ± 50 |
| Compound | 15i | SAHA | Compound | 15i | SAHA |
|---|---|---|---|---|---|
| HDAC1 | 217 ± 11 | 4.37 ± 0.24 | HDAC6 | 5.50 ± 0.23 | 7.80 ± 0.57 |
| HDAC2 | 398 ± 14 | 12.1 ± 1.28 | HDAC7 | 2600 ± 135 | >50000 |
| HDAC3 | 503 ± 25 | 3.34 ± 0.27 | HDAC8 | 1050 ± 98 | 1033 ± 62.5 |
| HDAC4 | >50000 | >50000 | HDAC11 | 2800 ± 161 | 895 ± 71.6 |
| HDAC5 | >50000 | >50000 |
| Kinase | Inhibition rate% | IC50a (nM) |
|---|---|---|
| GSK3 | 80% | 88 ± 2.7 |
| GSK3 | 82% | 69 ± 3.0 |
| CDK4/cyclin D1 | 17% | / |
| CDK6/cyclin D1 | 11% | / |
| FLT3 | 8% | / |
| BCR-ABL | 10% | / |
| EGFR | 5% | / |
| Compd. | IC50a | Compd. | IC50 |
|---|---|---|---|
|
| 1.62 ± 0.07 |
| 0.94 ± 0.02 |
|
| 8.02 ± 0.30 |
| 6.34 ± 0.26 |
|
| 9.50 ± 0.55 |
| 1.92 ± 0.09 |
|
| 2.55 ± 0.09 |
| 3.83 ± 0.16 |
|
| 1.96 ± 0.15 |
| 1.73 ± 0.20 |
|
| 1.29 ± 0.11 |
| 0.80 ± 0.04 |
| Tumour type | Cell line | 15i | SAHA. | ACY1215 |
|---|---|---|---|---|
| Cervical cancer | Hela | 1.22 ± 0.06 | 1.16 ± 0.05 | 10.40 ± 0.42 |
| Multiple myeloma | RPMI-8226 | 0.28 ± 0.01 | 0.43 ± 0.02 | 1.50 ± 0.14 |
| Liver cancer | HepG-2 | 1.82 ± 0.08 | 4.18 ± 0.29 | 14.42 ± 1.18 |
| Colon cancer | HT29 | 2.34 ± 0.08 | 2.63 ± 0.09 | 9.85 ± 0.25 |
| Breast cancer | MCF-7 | 0.69 ± 0.07 | 0.31 ± 0.02 | 6.24 ± 0.56 |
| Leukemia | OCI-AML3 | 0.41 ± 0.01 | 0.64 ± 0.03 | 5.24 ± 0.04 |
- —National Natural Science Foundation of China10.13039/501100001809
- —Natural Science Basic Research Program of Shaanxi Province10.13039/501100017596
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHistone Deacetylase Inhibitors Research · Cholinesterase and Neurodegenerative Diseases · Nuclear Receptors and Signaling
Introduction
Histone deacetylases (HDACs), which control the acetylation levels of nuclear proteins and cytoplasmic proteins, have been identified as promising targets for cancer therapy1–4. The classical 11 zinc-dependent isoforms comprise class I (HDAC1, HDAC2, HDAC3, HDAC8), class IIa (HDAC4, 5, 7, 9), class IIb (HDAC6, 10), and class IV (HDAC11)5^,^6. The anticancer mechanism of HDAC inhibition generally included reduced cell motility/migration, invasion, induction of apoptosis, angiogenesis, and blocking of DNA repair. HDAC6, which is primarily localised in the cytoplasm, exhibits distinct characteristics7^,^8. It features two tandem catalytic domains and acts directly on a host of cytosolic proteins and substrates such as α- and β-tubulin, heat shock protein, assembled micro-tubules and cortactin9–11, thereby regulating processes central to tumorigenesis. Besides, the binding of ubiquitin by unique zinc-finger domain enables HDAC6 regulate protein clearance and degradation. The favourable safety profile and reduced toxicity makes the development of HDAC6 inhibitor (HDAC6i) become a hot research in cancer treatment7^,^12. The pharmacophore of HDACis is well-established: a capping motif occupying the outside of the protein’s active pocket, a zinc-binding group (ZBG) chelating the catalytically active zinc ion, and a linker chain connecting the above two parts (Figure 1)13–17. Such a pharmacophore model usually applies to all isoforms due to the highly conserved nature of HDAC family18–20. Hydroxamic acid is the most commonly used ZBG, exemplified by approved pan-HDACis such as vorinostat (1, SAHA)21, belinostat (2)22 or panobinostat (3)23, and clinical selective HDAC6i such as ACY-1215 (4)24, ACY-241 (5)25 or KA2507 (6)26.
(A) Approved non-selective hydroxamic acid-based HDAC inhibitors; (B) clinical HDAC6 inhibitors.
Glycogen synthase kinase-3 (GSK-3) is an evolutionarily conserved serine/threonine kinase with an important role in various cellular processes, including cell proliferation, differentiation, apoptosis, and metabolism27^,^28. GSK3 has two distinct isoforms in mammals, α and β, which share high homology within their internal kinase domain29. Both GSK-3 isoforms are implicated in various cancer types, including colorectal, breast, prostate, pancreatic, and haematologic malignancies30. In particular, GSK3β is involved in multiple signal pathway including Wnt/β-catenin, PI3K/PTEN/AKT and Notch. It also functions in DNA repair through phosphorylation of DNA repair factors and affecting their binding to chromatin31. Thus far, several GSK-3 inhibitors have entered clinical trials such as Tideglusib (7)32, Elraglusib (8)33, and LY2090314 (9)34, as shown in Figure 2. Notably, Elraglusib had demonstrated encouraging efficacy and favourable safety against pancreatic cancer and other solid tumours in Phase II clinical trials.
Representative clinical GSK3 inhibitors.
The connection between HDACs and GSK3β have been highlighted35–38. Recent studies suggested the importance of co-inhibiting GSK3β and HDAC in cancers. Edderkaoui et al. demonstrated that a dual GSK3/HDAC inhibitor killed pancreatic cancer cells synergistically and suppressed pancreatic tumour growth and metastasis in mice39. Taylan et al. also displayed that dual inhibition of GSK3β and HDACs exerted significant antitumor effects in an ovarian cancer mouse model40. Meanwhile, the structural flexibility of HDAC inhibitors made them feasible for hybridisation with other inhibitors, a design strategy supported by numerous cases41–47. Consequently, developing dual GSK3/HDAC inhibitors was a rational and viable drug discovery.
Compound design
The common pharmacophore-based design strategy for multi-target drugs typically includes conjugated, fused, and merged pharmacophores48. 1,3,4-oxadiazole derivatives were reported for their anti-tumour and antioxidant properties49–51. In our design, the merged-pharmacophore design for de novo GSK3/HDAC dual-inhibitor was inspired by detailed examination of the binding mode of compound 14i (a known GSK3 inhibitor52) with GSK-3β (Figure 3). Its oxadiazole-phenyl scaffold occupied the adenosine-binding pocket, with one nitrogen atom forming a hydrogen bond with the protein and one oxygen atom from the dihydrodioxine ring interacting with hinge region. Both phenyl moieties engaged in hydrophobic interactions with the surrounding residues. The ester group did not provide any further interaction with the enzyme, as it was oriented towards the solvent. On the other hand, phenylhydroxamic acid group was a well-established and widely utilised pharmacophore in HDAC6i design, as shown in Tubastatin A (10, a highly selective HDAC6i53). Consequently, the direct conversion of the ester group in compound 14i to a hydroxamate afforded a novel inhibitor with potential dual activity against HDAC6 and GSK3. Here, we reported the synthesis, structure–activity relationship (SAR) study and antiproliferative evaluation of these merged oxadiazole derivatives.
Design of oxadiazole-based GSK3/HDAC6 dual inhibitors.
Chemistry
The primary modification site for these oxadiazole derivatives was focused on the capping phenyl. Compounds 15a–i were synthesised according to Scheme 1. Briefly, starting materials 11a–i were treated with hydrazine hydrate to provide the corresponding hydrazides 12a–i. Subsequent cyclisation of 12a–i with carbon disulphide yields the key intermediate 13a–i, as reported in relevant literatures52^,^54. Then, 13a–i reacted with methyl p-bromomethylbenzoate under mild conditions via nucleophilic substitution to form intermediates 14a–i. Direct aminolysis of intermediates 14a–i was precluded by concomitant oxadiazole ring-opening. Consequently, an alternative one-pot, three-step procedure was adopted. This involved hydrolysis of 14a–i to the corresponding carboxylic acids, followed by condensation with O-(tetrahydro-2H-pyran-2-yl)hydroxylamine and subsequent deprotection to furnish the target products 15a–i in higher overall yield.
Reagents and conditions: (a) NH2NH2.H2O, EtOH, 78 °C, overnight; (b) CS2, Et3N, EtOH, reflux, 8–12 h; (c) benzyl halides, 1 N NaOH, anhydrous DMF, r.t., 6–12 h; (d) NaOH, MeOH/H2O, r.t., overnight; (e) HATU, DIPEA, DMF, O-(tetrahydro-2H-pyran-2-yl) hydroxylamine, 0 °C, 6 h; (f) p-TsOH.H2O, MeOH, r.t., overnight.
Results and discussion
HDAC6, GSK3β activities and SAR study of target compounds
GSK3β and HDAC6 inhibitory activities of all compounds were evaluated with selective HDAC6is ACY1215 and Tubastatin A, non-selective SAHA, 13i and Elraglusib as the positives (Table 1). All nine compounds showed higher GSK3β activity than Elraglusib with IC_50_ in nanomolar level. 15h and 15i demonstrated the most potent inhibition with IC_50_ values of 88 nM and 95 nM, respectively. Replacing the 3, 4-methylenedioxy group with other substituents such as halogens or hydrogen resulted in diminished potency. The conversion of the ester to the hydroxamic acid led to a slightly decline of GSK3β activity (15i vs 14i), but all compounds displayed significant potency towards HDAC6. And seven of them exhibited single-digit nanomolar inhibitory activity against HDAC6. 15i emerged as the most potent compound with IC_50_ of 5.5 nM, comparable to that of ACY1215. For 15e, 15h and 15i, a polar oxygen atom on the capping phenyl enhanced HDAC6 activity compared to halogen substituents or unsubstituted phenyl analog. Based on the above results, it was noteworthy that the substituent effects on the capping phenyl exhibited a favourable alignment between HDAC6 and GSK3β activity. And 15i was identified as the preferred candidate for further investigation.
15i was evaluated for isoform selectivity against other HDACs. As depicted in Table 2, 15i inhibited HDAC1 with IC_50_ of 217 nM, leading to a 40∼fold selectivity index. Besides, 15i also showed moderate inhibition against HDAC2 and 3, weak activity against HDAC7 and 8, and no activity against HDAC4 and 5. In kinase panel screening, 15i exhibited comparable inhibitory activity against GSK-3α (IC_50_ = 69 nM). It was also tested against five common kinases such as cyclin-dependent kinase 4 (CDK4)/cyclin D1, CDK6/cyclin D1, Fms-like tyrosine kinase 3 (FLT3), breakpoint cluster region-Abelson (BCR-ABL) and epidermal growth factor receptor (EGFR). As shown in Table 3, 15i showed weak off-target effects against the profiled kinases.
Molecular simulation
Due to the high sequence identity in the active pockets of GSK3α and GSK3β, GSK3β was chosen for molecular docking study because its role in tumorigenesis was more extensively characterised. Compound 15i was docked into human GSK3β and HDAC6 proteins to validate the SAR underlying the enzymatic activity. As outlined in Figure 4A, the phenyl-oxadiazole core of 15i located in GSK3β protein’s adenine-binding site and retained two critical hydrogen bond with the residues of Lys85 and Val135, which was also observed in the binding mode of GSK3β with 14i. The incorporation of hydroxamic acid group did not significantly alter the binding mode of 15i. The interaction between 15i and HDAC6 was displayed in Figure 4B, hydroxamate chelated with Zn^2+^ in a bidentate manner with Zn^2+^–O distances of 2.5 Å and 1.4 Å for the OH and C=O groups, respectively. The residue of Gly619 additionally accepted a hydrogen bond from hydroxamate group. The phenyl linker of 15i embed into the channel between Phe620 and Phe680. Meanwhile, oxadiazole scaffold formed two H-bonds with Ser568 and enabled the capping group to align well with the amino acids on the rim of the binding tunnel. Dihydrodioxine in capping group oriented towards the polar solvent and an oxygen atom interacted with Asn494 by H-bond. Hence, dihydrodioxine group played an important role in binding to both proteins, which rationalises the high enzymatic activity of 15i. Besides, 15i was also docked into HDAC1 protein to rationalise the isoform selectivity of these oxadiazole derivatives. As illustrated in Figure 4C, hydroxamic acid group of 15i also coordinated with zinc ion at the bottom of HDAC1 protein and formed one H-bond with His140. However, the oxadiazole core in cap region failed to form additional H-bonds with HDAC1 in contrast to its binding with HDAC6. Moreover, the distances between hydroxamic acid and zinc ion were greater than those in HDAC6 complex, which indicated a potentially weaker chelation.
(A) Binding model of 15i (yellow) in the catalytic pocket of GSK-3β (PDB code: 3F88). (B) Binding model of 15i (yellow) in the catalytic pocket of human HDAC6 (PDB code: 5EDU). (C) Binding model of 15i (yellow) in the catalytic pocket of human HDAC1 (PDB code: 5ICN). GSK3β protein, HDAC6 protein and HDAC1 was labelled in burgundy, green and light blue, respectively. The hydrogen bonds were labelled in blue. Zinc ion was shown in brown.
Western blot assay
To further confirm the intracellular target specificity of 15i, gastric cancer cell line AGS were treated with 15i at concentrations of 1, 5, and 10 μM, along with HDAC6i Tubastatin A (a highly selective HDAC6i) and SAHA at 10 μM as control groups. Selective HDAC6i generally upregulated acetylated α-tubulin (Ac-α-tubulin) expression with minimal effects on acetylated histone H3 (Ac-H3) level15^,^55. As shown in Figure 5A and B, 15i dose-dependently increased acetylated α-tubulin level while only inducing moderate Ac-H3 expression. In contrast, the pan-inhibitor SAHA significantly elevated both Ac-α-tubulin and Ac-H3 levels.
*(A). Effect of compound 15i on the acetylation of histone H3 and α-tubulin in AGS cell line (n = 3); (B) Densitometry analysis of protein Ac-H3 and Ac-a-tubulin. Data are the average of three independent experiments mean ± SD; **p < 0.01, **p < 0.001 indicated comparison with the control group.
Antiproliferative activity and mechanism study
Given that gastric cancer is one of the most common and deadly cancers globally56, all derivatives were evaluated against gastric cancer cell line AGS in initial in vitro screening. As shown in Table 4, all nine compounds demonstrated potent antiproliferative activity with IC_50_ values within single-digit micromolar range, better than ACY-1215 and 14i. Compared to ACY-1215, pan-inhibitor SAHA showed superior antiproliferative activity (IC_50_ = 1.62 μM), which is partially attributed to its non-selective inhibition of Class I HDACs57. While dual inhibitors 15b, 15c, 15d, 15f and 15h showed comparable activity to SAHA with IC_50_s between 1.2 9 and 1.96 μM. Significantly, 15i showed the highest potency (IC_50_ = 0.80 μM) yet low activity against HDAC1-3, indicated a synergistic antiproliferative effect from dual GSK3/HDAC6 inhibition. Based on these findings, we subsequently performed a broad-spectrum antitumor screening for compound 15i. As shown in Table 5, 15i was evaluated against different cancer cell lines including Hela, RPMI-8226, HepG-2, HT29, MCF-7 and OCI-AML3. All these cells were sensitive to HDAC inhibitors. 15i demonstrated submicromolar activity against three cell lines RPMI-8226, MCF-7 and OCI-AML3 with IC_50_ values of 0.28, 0.69 and 0.41 μM, respectively. And 15i also showed single-digit micromolar activity against three others with IC_50_s range from 1.22 to 4.22 μM. The potency was comparable to SAHA and significantly superior to that of ACY-1215. In flow cytometry assay, as expected, 15i dose-dependently induced AGS cell apoptosis (Figure 6). An apoptosis rate of 85.93% incubated with 15i at 4 μM was significantly higher than that of ACY1215 (26.25%), comparable to SAHA (88.3%). The pro-apoptotic activity of compound 15i was further validated by DNA-binding dye Hoechst 33342 assay (Figure 7). Mechanistic study demonstrated that 15i downregulated the expression of caspase-3, caspase-9, and PARP in a dose-dependent manner, thereby inducing apoptosis (Figure 8).
*(A) Induction of apoptosis at 48 h by compound 15i at 1, 2, 4 μM concentration in AGS cell line by flow cytometry analysis with SAHA (4 μM) and ACY1215 (4 μM) as controls. The percentage of cells in each part was indicated. (B) The histogram of SAHA, ACY-1215 and 15i on AGS cells apoptosis. Data are the mean ± SD of three independent experiments; *p < 0.05, **p < 0.01, **p < 0.001 indicated comparison with the control group.
*The effect of 15i on AGS cell apoptosis at 4 μM was determined by Hoechst 33342 staining with SAHA (4 μM) and ACY1215 (4 μM) as controls. Data were subjected to one-way ANOVA. Bars indicate densitometric analysis of immunoblots. Data are the mean ± SD of three independent experiments: *p < 0.05, **p < 0.01, **p < 0.001 compared with the control group.
*Western blot assay of 15i on the proteins expression of caspase3, caspase9 and PARP at 0.5, 1, and 2 μM, respectively. The chart showed the densitometric analysis of caspase-3, caspase-9, and PARP expression from Western blots. Data were the average of three independent experiments mean ± SD; p < 0.05, ** p < 0.01, *** p < 0.001 indicated comparison with the control group.
Conclusion
Given the complexity of tumorigenesis, a series of dual-target inhibitors were designed based on the biological synergy between GSK3 kinase and HDAC6 in our research. Unlike common design for kinase/HDAC dual inhibitors that fused two pharmacophores together via a linker, we utilised an oxadiazole core to elegantly merge key pharmacophores with maximal overlap. The resulting lean molecular structure was a highlight of this study. Enzymatic assays confirmed that all synthesised derivatives exhibited potent HDAC6 inhibition and moderate activity against GSK3. Screening against other relevant targets showed that this class of compounds had minimal off-target activity. Cell-based assays demonstrated that these dual-target inhibitors exhibited significantly better antiproliferative effect against a panel of cancer cell lines than clinical HDAC6i ACY-1215. These results indicated these dual GSK3/HDAC6 inhibitors hold greater potential than single-target inhibitors. Currently, further structural modifications and in vivo evaluation are underway.
Experimental section
Chemistry
All of the starting reagents were purchased and were used with no additional purification. All of the mentioned yields were for isolated products. Melting points were determined in open capillaries on a WRS-1A digital melting point apparatus (Shenguang).^1^H-NMR spectras were detected on a Bruker DRX–400 (400 MHz) using TMS as internal standard. High resolution mass spectra were obtained from Thermo Scientific Q Exactive. The chemical shifts were reported in ppm (δ) and coupling constants (J) values were given in Hertz (Hz). The purities of all target compounds were tested by HPLC to be > 95.0%. HPLC analysis was performed at room temperature using an Agilent Eclipse XDB-C18 (250 mm × 4.6 mm) and plotted at 254 nm by 30% MeOH/H_2_O as a mobile phase.
General procedure for the synthesis of compounds 15a–i
Compounds 14a–i were synthesised according to the standard procedure in literature52. Then, 15a–i were synthesised by one-pot, three-step synthesis: (i) To a solution of various esters 14a–i (1 equiv) in methanol and H_2_O (v/v = 1: 1) was added NaOH (3 equiv) The mixture was stirred at room temperature. The reaction was monitored by TLC until the maximum conversion. After neutralised with acetic acid, the formed precipitate was collected by filtration, washed with water, and dried in vacuo to give compounds for next step; (ii) At 0 °C, compounds (1 equiv) from step (i) were added to a solution of DIPEA (4 equiv) and HATU (1.1 equiv) in DMF. After 30 min, O-(tetrahydro-2H-pyran-2-yl)hydroxylamine (1 equiv) was added. The mixture was stirred for additional 4–6 h. After Then, the mixture was poured into water and extracted with EtOAc. The combined organic extracts were washed with brine, and concentrated in vacuo; (iii) To a solution of compounds (1 equiv) from step (ii) in methanol was added TsOH^.^H_2_O (0.2 equiv), and the mixture was stirred at room temperature for overnight. After removal of volatiles, residues were purified by chromatography on a silica gel column.
N-hydroxy-4-(((5-phenyl-1, 3, 4-oxadiazol-2-yl)thio)methyl)benzamide (15a)
Yellow solid, yield 69.6%, m.p.: 180.3–181.9 °C.^1^H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 9.03 (s, 1H), 7.95 (dd, J = 8.0, 1.5 Hz, 2H), 7.71 (d, J = 8.3 Hz, 2H), 7.64 − 7.54 (m, 5H), 4.62 (s, 2H). ^13^C NMR (101 MHz, DMSO-d6) δ 165.35, 163.89, 163.20, 140.01, 132.20, 132.13, 129.51, 129.10, 127.18, 126.45, 122.99, 35.44. HR-MS (ESI, m/z): Calcd for 328.07504. (C_16_H_14_N_3_O_3_S^+^ [M + H]^+^). Found 328.07462.
N-hydroxy-4-(((5–(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)thio)methyl)benzamide (15b)
Yellow solid, yield 67.3%, m.p.: 201.3–203.1 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 9.03 (s, 1H), 8.17 (d, J = 8.2 Hz, 2H), 7.97 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 4.65 (s, 2H). ^13^C NMR (101 MHz, DMSO-d6) δ 164.29, 164.16, 163.87, 139.87, 132.23, 131.72, 131.40, 129.10, 127.28, 127.18, 126.75, 126.44, 126.40, 125.08, 122.37, 35.42. HR-MS (ESI, m/z): Calcd for 396.06242. (C_17_H_13_F_3_N_3_O_3_S^+^ [M + H]^+^). Found 396.06210. (C_17_H_13_F_3_N_3_O_3_S^+^ [M + Na]^+^). Found 418.04404.
4-(((5-(4-chlorophenyl)-1,3,4-oxadiazol-2-yl)thio)methyl)-N-hydroxybenzamide (15c)
Yellow solid, yield 60.3%, m.p.:178.5–180.1 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 9.03 (s, 1H), 7.96 (d, J = 8.6 Hz, 2H), 7.69 (dd, J = 17.2, 8.4 Hz, 4H), 7.55 (d, J = 8.2 Hz, 2H), 4.62 (s, 2H). ^13^C NMR (101 MHz, DMSO-d6) δ 164.58, 163.87, 163.47, 139.93, 136.79, 132.20, 129.64, 129.08, 128.24, 127.16, 121.87, 35.42. HR-MS (ESI, m/z): Calcd for 384.01801. (C_16_H_12_NaCl^35^N_3_O_3_S^+^ [M + Na]^+^). Found 384.01779.
N-hydroxy-4-(((5–(4-methoxyphenyl)-1,3,4-oxadiazol-2-yl)thio)methyl)benzamide (15d)
White solid, yield 65.6%, m.p.: 153.2–154.9 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H), 9.03 (s, 1H), 7.89 (d, J = 8.9 Hz, 2H), 7.70 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 8.9 Hz, 2H), 4.60 (s, 2H), 3.85 (s, 3H). ^13^C NMR (101 MHz, DMSO-d6) δ 165.29, 164.01, 163.88, 162.32, 162.11, 158.62, 142.64, 140.05, 132.16, 129.05, 128.31, 127.15, 115.31, 114.93, 55.57, 35.46. HR-MS (ESI, m/z): Calcd for 380.06755. (C_17_H_15_NaN_3_O_4_S^+^ [M + Na]^+^). Found 380.06705.
4-(((5–(4-fluorophenyl)-1,3,4-oxadiazol-2-yl)thio)methyl)-N-hydroxybenzamide (15e)
White solid, yield 63.5%, m.p.: 189.7–191.4 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 9.04 (s, 1H), 8.03 (dd, J = 8.9, 5.3 Hz, 2H), 7.72 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.2 Hz, 2H), 7.45 (t, J = 8.9 Hz, 2H), 4.63 (s, 2H). ^13^C NMR (101 MHz, DMSO-d6) δ 165.39, 164.60, 163.88, 163.20, 139.96, 132.19, 129.23, 129.14, 129.07, 127.16, 116.85, 116.63, 35.43. HR-MS (ESI, m/z): Calcd for 368.04756. (C_16_H_12_NaFN_3_O_3_S^+^ [M + Na]^+^). Found 368.04724.
4-(((5–(3, 4-dichlorophenyl)-1,3,4-oxadiazol-2-yl)thio)methyl)-N-hydroxybenzamide (15f)
Yellow solid, yield 64.8%, m.p.: 183.6–185.1 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 9.04 (s, 1H), 8.15 (d, J = 1.9 Hz, 1H), 7.93 (dd, J = 8.4, 2.0 Hz, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.2 Hz, 2H), 4.64 (s, 2H). ^13^C NMR (101 MHz, DMSO-d6) δ 163.98, 163.65, 139.93, 134.78, 132.38, 132.21, 131.87, 129.12, 128.12, 127.17, 126.54, 123.48, 35.38. HR-MS (ESI, m/z): Calcd for 417.9790. (C_16_H_11_NaCl_2_N_3_O_3_S^+^ [M + Na]^+^). Found 417.9787.
4-(((5–(3-chlorophenyl)-1,3,4-oxadiazol-2-yl)thio)methyl)-N-hydroxybenzamide (15 g)
Yellow solid, yield 66.9%, m.p.: 182.1–183.8 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 11.20 (s, 1H), 9.05 (s, 1H), 7.97 − 7.91 (m, 2H), 7.71 (d, J = 8.2 Hz, 3H), 7.63 (t, J = 7.9 Hz, 1H), 7.56 (d, J = 8.2 Hz, 2H), 4.63 (s, 2H).^13^C NMR (101 MHz, DMSO-d6) δ 164.23, 163.78, 139.97, 134.12, 132.21, 131.92, 131.55, 129.12, 127.18, 125.99, 125.15, 124.93, 35.39. HR-MS (ESI, m/z): Calcd for 384.01801. (C_16_H_12_NaCl^35^N_3_O_3_S^+^ [M + Na]^+^). Found 384.01755.
4-(((5–(2, 3-dihydrobenzo[b][1,4]dioxin-6-yl)-1,3,4-oxadiazol-2-yl)thio)methyl)-N-hydroxybenzamide (15h)
White solid, yield 58.6%, m.p.: 176.2–177.7 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 11.20 (s, 1H), 9.05 (s, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 7.44 − 7.39 (m, 2H), 7.05 (d, J = 8.4 Hz, 1H), 4.59 (s, 2H), 4.32 (d, J = 4.0 Hz, 4H). ^13^C NMR (101 MHz, DMSO-d6) δ 165.00, 163.86, 162.49, 146.76, 143.86, 140.03, 132.15, 129.03, 127.13, 119.99, 118.20, 115.91, 115.09, 64.46, 64.09, 35.41. HR-MS (ESI, m/z): Calcd for 394.04681. (C_18_H_16_N_3_O_5_S^+^ [M + Na]^+^). Found 394.04603.
4-(((5-(Benzo[d][1, 3]dioxol-5-yl)-1,3,4-oxadiazol-2-yl)thio)methyl)-N-hydroxybenzamide (15i)
Yellow solid, yield 62.9%, m.p.: 186.5–188.2 °C. ^1^H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H), 9.02 (s, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.56 − 7.48 (m, 3H), 7.44 (d, J = 1.6 Hz, 1H), 7.11 (d, J = 8.2 Hz, 1H), 6.16 (s, 2H), 4.60 (s, 2H). ^13^C NMR (101 MHz, DMSO-d6) δ 165.15, 163.87, 162.51, 150.51, 148.16, 140.04, 132.17, 129.06, 127.15, 121.77, 116.63, 109.20, 106.18, 102.18, 35.41. HR-MS (ESI, m/z): Calcd for 408.06246. (C_17_H_14_N_3_O_5_S^+^ [M + Na]^+^). Found 408.06201.
In vitro HDAC enzyme assay
IC_50_ testing of compounds were performed by Reaction Biology Corporation. Detailed methods can be found in our previously published literature43.
Kinase inhibition assay
IC_50_ testing of compounds was performed by Reaction Biology Corporation. Detailed methods can be found in https://www.reactionbiology.com/services/biochemical-assays/kinase-screening/.
Cell culture and antiproliferative assay
All cells (Procell Life Science and Technology Co., Ltd) were maintained at 37 °C in a humidified atmosphere of 5% CO_2_ in air. The cells were cultured in IMDM medium with 20% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin. Briefly, 100 µL cell suspension or completed medium were plated into 96-well plate (5 × 10^4^ cells per well) for 24 h. The compounds were serially diluted to concentrations of 20, 10, 5, 2.5, 1.25, 0.625 0.313 μM and incubated for 48 h; Then, Alamar blue solution (10%v/v) were pipetted into each well of 96-well plate; and the plate was incubated for an additional 5–6 h. The absorbance (OD) was read at 530/590 nm. Data were normalised to vehicle groups (DMSO) and represented as the means of three independent measurements with standard errors of <20%. The IC_50_ values were calculated using Prism 5.0.
Apoptosis assay
For flow cytometry assay, detailed methods can be found in our previously published literature43.
For Hoechst 33342 staining assay, 1.5 ml AGS cells (3.0 × 10^5^ cells/mL) were seeded and incubated for 24 h, Then, the control and experimental groups were supplemented with 1.5 ml of fresh medium containing 0.1% DMSO and compounds at 4 μM, respectively. After incubated for 48 h, 750 μL Hoechst 33324 (2 μg/mL) was added to each dish and incubated for 20 min. Then, the samples were washed with PBS and examined for specific staining using a laser scanning confocal microscope at 405 nm.
Western blotting assay
AGS (purchased from Procell Life Science and Technology Co., Ltd; NO. PC-H2023011119; 3 × 10^5^) were seeded overnight and incubated with compound 15i for 48 h on indicated concentrations. Cell extract was prepared by lysing cultured cells with a mammalian protein extraction reagent supplemented with EDTA-free protease inhibitor for 15 min. Supernatants were collected following centrifugation of lysed cells at 15000g for 10 min at 4 °C. Protein samples (15 μg per lane) were resolved by SDS-PAGE and subsequently transferred to PVDF membranes (0.45 μM). Membranes were blocked with 5% non-fat milk in TBST and subsequently incubated overnight at 4 °C with primary antibodies against Ac-H3 (abcom, AB32129) and Ac-ɑ-tubulin (Cell Signalling, 2144), PARP, Caspase-9, Caspase-3, and GAPDH. After incubation with a rabbit-derived secondary antibodies, immunoreactive bands were visualised by chemiluminescence. Quantification was performed using ImageJ (v2.0), and data were analysed with GraphPad Prism (v8.0).
Computational methods
Molecular docking was performed using Sybyl-X 2.0 software (222 S Central Ave Ste 1008, Saint Louis, MO 63105, USA) based on the cocrystal of HDAC6 (PDB: 5EDU). The cavity occupied by trichostatin A was selected as the ligand binding site. For HDAC1, the cocrystal of PDB: 5ICN was used for docking. GLY-ALA-6A0-ARG-HIS was selected as the ligand binding site. For GSK3β, the cocrystal of PDB: 3F88 was used for docking. And 5–(1-(4-methoxyphenyl)-1H-benzimidazol-6-yl)-1, 3, 4-oxadiazole-2(3H)-thione was selected as the ligand binding site. Water molecules outside the binding pocket were excluded. The other docking parameters were kept as default.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shirvaliloo M. The landscape of histone modifications in epigenomics since 2020. Epigenomics. 2022;14(23):1465–1477.36710634 10.2217/epi-2022-0437 · doi ↗ · pubmed ↗
- 2Zhang Y, Sun Z, Jia J, Du T, Zhang N, Tang Y, Fang Y, Fang D. Overview of histone modification. Adv Exp Med Biol. 2021;1283:1–16.33155134 10.1007/978-981-15-8104-5_1 · doi ↗ · pubmed ↗
- 3Gediya P, Parikh PK, Vyas VK, Ghate MD. Histone deacetylase 2: a potential therapeutic target for cancer and neurodegenerative disorders. Eur J Med Chem. 2021;216:113332.33714914 10.1016/j.ejmech.2021.113332 · doi ↗ · pubmed ↗
- 4Cheng B, Pan W, Xiao Y, Ding Z, Zhou Y, Fei X, Liu J, Su Z, Peng X, Chen J. HDAC-targeting epigenetic modulators for cancer immunotherapy. Eur J Med Chem. 2024;265:116129.38211468 10.1016/j.ejmech.2024.116129 · doi ↗ · pubmed ↗
- 5de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDA Cs): characterization of the classical HDAC family. Biochem J. 2003;370(Pt 3):737–749.12429021 10.1042/BJ 20021321 PMC 1223209 · doi ↗ · pubmed ↗
- 6Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338(1):17–31.15050820 10.1016/j.jmb.2004.02.006 · doi ↗ · pubmed ↗
- 7Kalin JH, Bergman JA. Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J Med Chem. 2013;56(16):6297–6313.23627282 10.1021/jm 4001659 · doi ↗ · pubmed ↗
- 8Lo Presti P. HDAC 6 in diseases of cognition and of neurons. Cells. 2020;10(1):12.33374719 10.3390/cells 10010012 PMC 7822434 · doi ↗ · pubmed ↗