Microbiome composition of Drosophila suzukii varies across geographical regions
Matthew J. Medeiros, Allexa D. Burger, Donald K. Price, Joanne Y. Yew

TL;DR
The study finds that the microbiome of the invasive fruit fly Drosophila suzukii varies by region, suggesting environmental factors shape its microbial community.
Contribution
The paper reveals that D. suzukii incorporates region-specific microbes while maintaining conserved bacterial families, aiding its adaptability.
Findings
Wild D. suzukii populations from different regions have distinct microbial compositions.
Seven bacterial families are conserved across all D. suzukii populations.
In Hawaiʻi, non-native D. suzukii has different bacterial communities from native species but similar fungal profiles.
Abstract
Drosophila suzukii is a common agricultural pest in numerous parts of the world, costing more than $500 million annually in crop loss in the United States alone. Understanding the genetic and physiological mechanisms underlying its remarkable adaptability has been a major focus for the agricultural industry as well as evolutionary biologists. The microbiome, the community of microbes associated with host organisms, can play a pivotal role in local adaptation by improving host resilience to environmental stress and providing access to new sources of nutrition. Here, we test the hypothesis that the colonization of nonnative regions is associated with the incorporation of regionally-specific microbial taxa. We compare the microbiome profiles of wild-caught D. suzukii across five global sites, Asia, Europe, the United Kingdom, North America, and Hawaiʻi. We also compare microbial…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
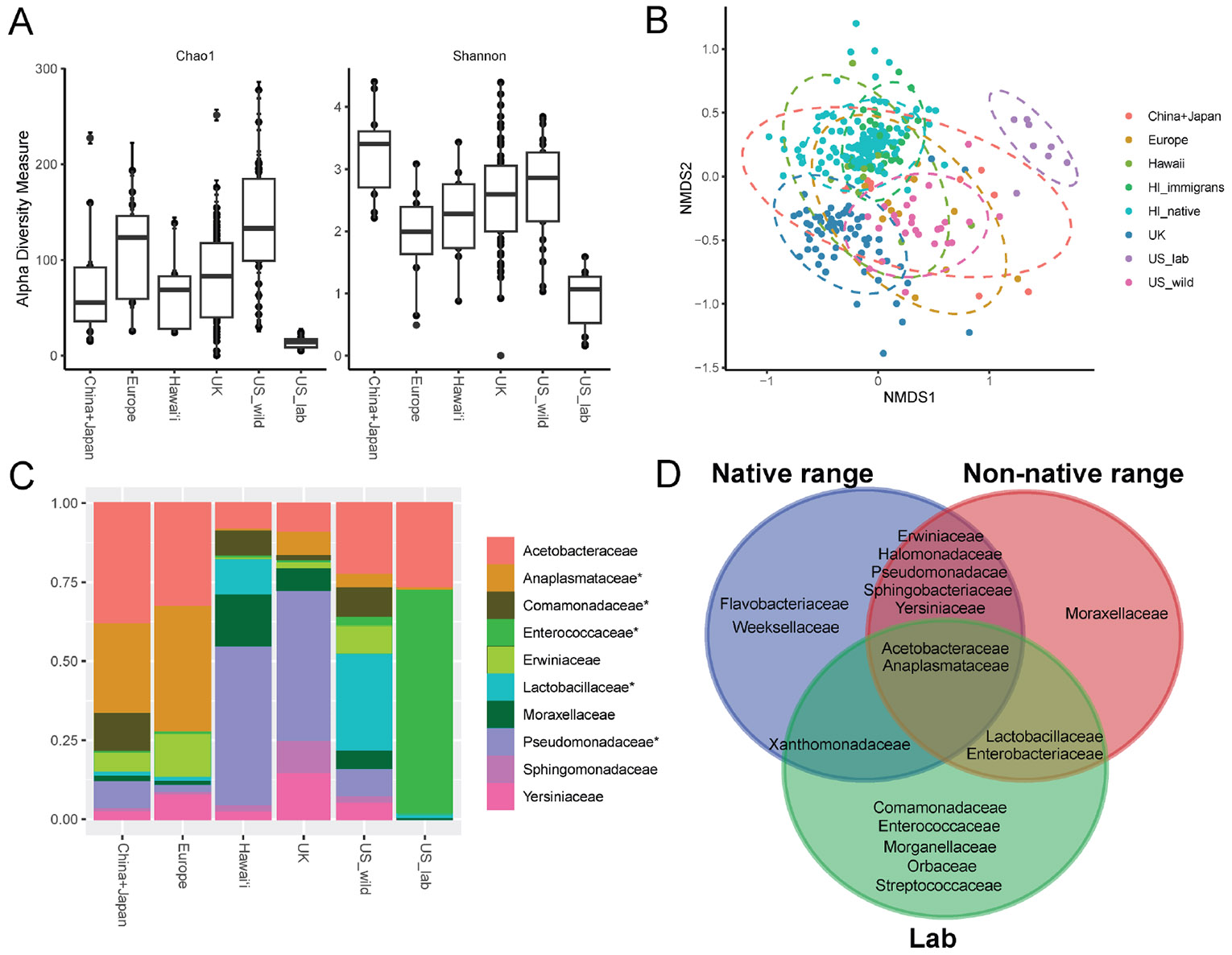 Figure 1
Figure 1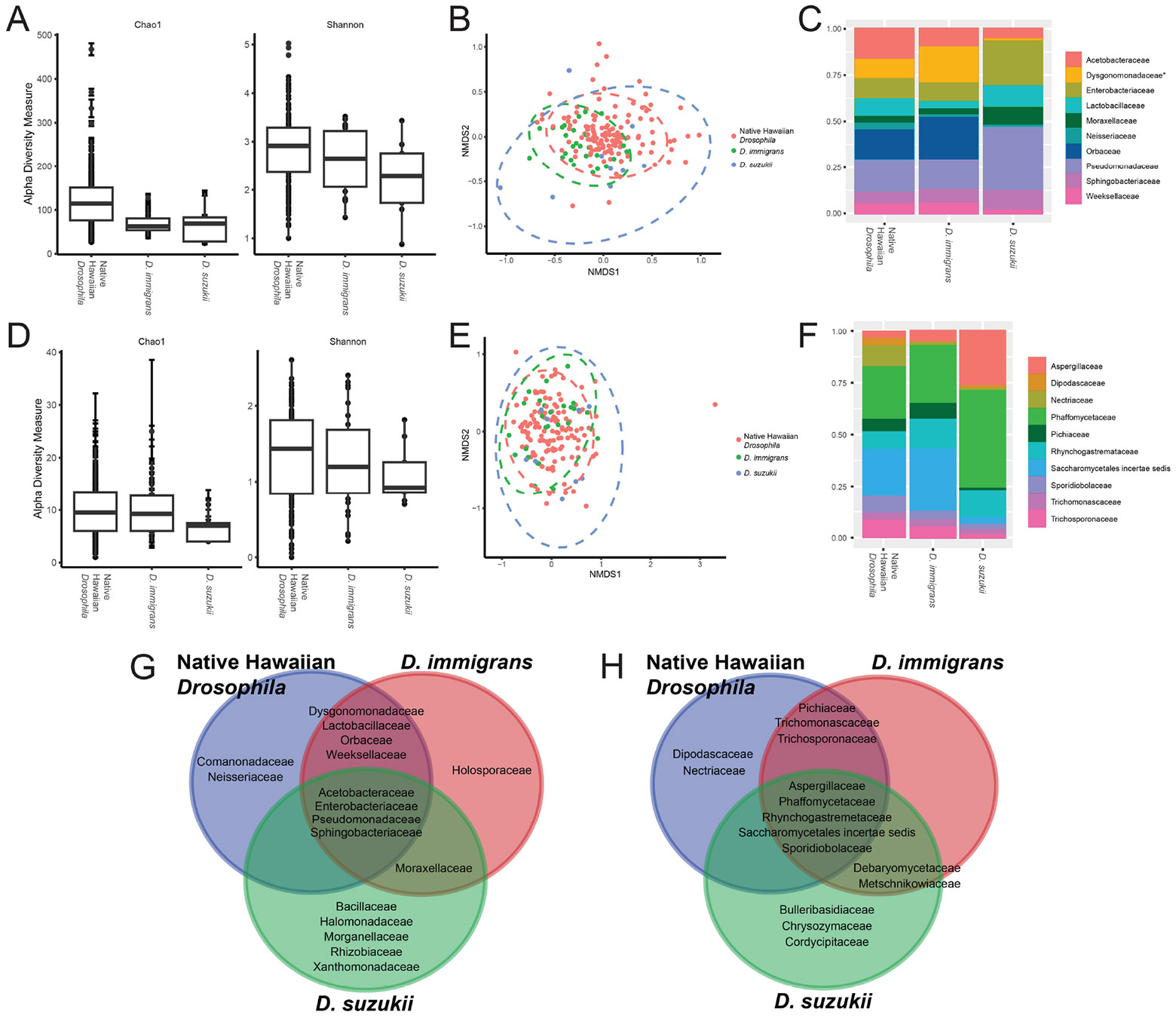 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect behavior and control techniques · Invertebrate Immune Response Mechanisms · Insect symbiosis and bacterial influences
Introduction
The spotted wing Drosophila, Drosophila suzukii, is considered a globally invasive pest, having spread from its native range in East Asia (Kanzawa, 1939; Bolda et al., 2010) to disparate locations in North America (Hauser, 2011), Europe (Calabria et al., 2012), South American (Deprá et al., 2014; Andreazza et al., 2017), North African countries (Kwadha et al., 2021), and Asia (Calabria et al., 2012; Cini et al., 2012). D. suzukii’s preference to oviposit in soft-flesh fruits has resulted in significant yield losses of fruit crops including cherries, grapes, and plums (Tait et al., 2021). The success of D. suzukii in expanding its range is partly attributed to its high adaptability to novel habitats and ecological niches (Poyet et al., 2015; Little et al., 2020). Within Hawaiʻi, the species is found across the islands of Kauaʻi, Oʻahu, Molokaʻi, Maui, and Hawaiʻi Island (Kaneshiro, 1983; Leblanc et al., 2009; Hauser, 2011) in lower elevation agricultural parks as well as high elevation native forest reserves. D. suzukii is capable of thriving in elevations over 2000 m, and at mean annual temperatures of less than 12 °C (Sánchez-Ramos et al., 2019a, b; Curbelo et al., 2022), near the lowest temperatures considered viable for activity in this genus (Koštál et al., 2016). In addition, D. suzukii has been observed feeding on a variety of native and non-native fruits (Magnacca et al., 2008; Koch et al., 2020). Previous genetic analyses of D. suzukii populations from Asia (Feng et al., 2024), Hawaiʻi (Koch et al., 2020), the continental United States (Mérel et al., 2021), Europe, and South America (Adrion et al., 2014) identified genetic divergences that may facilitate successful environmental adaptation. The microbiome has also been hypothesized as a major driver of local evolution allowing organisms to exploit new ecological niches by enhancing host physiology and behavior (Shu et al., 2021). Indeed, the ability to consume novel foods in a newly colonized area has been proposed as a key factor for successful invasion (Shik and Dussutour, 2020). D. suzukii uses microbes to aid metabolism and survival in their preferred high-sugar and low-protein fruit hosts (Bing et al., 2018; Gao et al., 2023) and may use naturally occurring microbes on a new host plant to extract essential nutrients (Lin et al., 2021). Local microbes may also aid in the tolerance of temperatures near the extreme of their ranges (Mueller et al., 2011; Chevalier et al., 2015; Houwenhuyse et al., 2021).
To address the possibility that the colonization of new ecological niches by D. suzukii is associated with changes in microbiome composition, we compared bacterial profiles of wild D. suzukii populations from the native ranges in China and Japan to populations from non-native ranges in Europe, the United Kingdom (UK), North America, and Hawaiʻi (Martinez-Sañudo et al., 2018) and lab-reared D. suzukii from the United States (US) (Bing et al., 2018; Martinez-Sañudo et al., 2018; Lin et al., 2021). To assess how closely D. suzukii microbiomes resembles that of other native and invasive species found in similar habitats, we compared Hawaiian D. suzukii bacterial and fungal profiles to another cosmopolitan drosophilid found in Hawaiʻi, D. immigrans, as well as to native Hawaiian picture-wing Drosophila.
Materials and methods
Sample collection of drosophilids in the Hawaiian Islands
D. suzukii, D. immigrans, and endemic Hawaiian drosophilidae were collected from the islands of Molokaʻi, Lanaʻi, and Hawaiʻi Island using sponges baited with mushrooms, banana, and yeast. Samples were immediately placed into 95% ethanol, transported on ice packs, and stored at −80 °C in the laboratory until processing. Metadata associated with samples collected in Hawaiʻi are provided in Supplementary Tables S1, S2.
Library preparation and sequencing of Hawaiian dosophilidae
Details of DNA extraction and library preparation are as previously described (Medeiros et al., 2025). Briefly, surface-sterilized flies were homogenized using a bead mill homogenizer (Bead Ruptor Elite, Omnic, Inc; GA, USA) and DNA extracted with PowerMag Bead Solution kit (Qiagen; MD, USA) according to manufacturer’s instructions. The 16S rRNA gene was amplified with primers to the V4 region (515F: GTGYCAGCMGCCGCGGTAA; 806R: GGACTACNVGGGTWTCTAAT) (Parada et al., 2016). Fungal diversity was characterized using primers to the internal transcribed spacer (ITS1f: CTTGGTCATTTAGAGGAAGTAA; ITS2: GCTGCGTTCTTCATCGATGC) (White et al., 1990). The primers contain a 12-base pair Golay-indexed code for demultiplexing. The PCRs were performed with the KAPA3G Plant kit (Sigma Aldrich, MO, USA) using the following parameters: 95 °C for 3 min, followed by 35 cycles of 95 °C for 20 seconds, 50 °C for 15 seconds, 72 °C for 30 seconds, and a final extension for 72 °C for 3 min. The PCR products were cleaned and normalized with the Just-a-plate kit (Charm Biotech, MO, USA). High throughput sequencing (HTS) was performed with MiSeq and 250 bp paired-end kits (Illumina, Inc., CA, USA).
Data processing of 16S rRNA amplicons for multi-region comparisons
Taxonomic analyses of D. suzukii, D. immigrans, and native Hawaiian Drosophila microbiomes were assessed from next-generation amplicon sequencing of regions in the 16S rRNA gene using DADA2 v1.16 (Callahan et al., 2016). All analyses were conducted using R version 4.4.2 (R Core Team, 2022). Four publicly accessible projects deposited on the NCBI Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra) and in-house data derived from D. suzukii, D. immigrans, and native Hawaiian Drosophila collected in Hawaiʻi by the authors of this study were used in the analysis for a total of five independent sources of sequencing data (Supplementary Table S3). FASTQ files deposited onto the SRA were retrieved using fasterqdump command, part of the NCBI SRA Toolkit. SRA projects included in this study are PRJEB50289 (Fountain et al., 2018), PRJNA347319 (Martinez-Sañudo et al., 2018), PRJNA412893, and PRJNA719706 (Lin et al., 2021). All raw sequence reads were demultiplexed before analysis.
All sequence data were generated from paired-end Illumina sequencing strategy, however there was no consensus in the primer sets used for all projects. Additionally, two of the SRA projects (PRJNA347319 and PRJNA719706) reported paired-end read strategy but only one spot contained read information from the SRA database; thus, only one file per sample was extracted. Visualization of the quality profile plots of these data revealed that the SRA data contained merged forward and reverse reads, referred to herein as extended fragments. Due to the unique nature of each dataset used in this study, each project was handled separately for pre-processing. A custom R script was used to search for the presence of sequencing primers in the reads. Sequencing primers were reported for all but SRA projects PRJNA412893, and in this case the primers used were inferred using the custom script to search for common 16S rRNA sequencing primers until the primer sites were identified. Locations of the primer sites in the forward and reverse reads were used to define the trimming (trimLeft) and the quality plots were used to define the truncation (truncLen) filtering parameters in DADA2. A default max EE setting of 2 was used for both forward and reverse reads, but one project required raising the threshold to recover enough sequence reads passing the filter. For extended fragments, as suggested by the DADA2 creator on github, the error rates were inflated by 3 to account for the heterogeneity between the merged subsegments using the command inflateErr. Project-specific details including primers, and pre-processing parameters are given in Supplementary Table S4.
After filtering, trimming and estimating error rates, paired-end reads were merged following the standard DADA2 workflow. Only merged reads or extended fragments (i.e., previously merged reads) were used for downstream analysis. The merged reads (and extended fragments) for 16S rRNA gene sequences from all five projects were used to create a sequence table, remove chimeras and assign taxonomy (excluding mitochondria and chloroplasts) using the SILVA SSU Ref NR database version 138.1 (Quast et al., 2013; Yilmaz et al., 2014). The number of reads tracked through the processing pipeline for each D. suzukii sample is given in Supplementary Table S5. Taxonomy assignments for 16S rRNA reads were based on amplicon sequence variant (ASV) data (100% similarity). Each sample was rarefied with a subsampling depth of 5,000 ASVs.
Data processing of ITS amplicons from Hawaiʻi drosophilidae samples
Post-processing of HTS data (filtering, trimming, and clustering) for Hawaiʻi samples (native Hawaiian flies, D. immigrans, D. suzukii) was performed using the “MetaFlow∣mics” Fungal ITS pipeline for fungi which uses the DADA2 workflow (Arisdakessian et al., 2020; Medeiros et al., 2024). ASV clustering was performed at the 97% similarity threshold. Taxonomy assignments for ITS reads were performed with NCBI BLAST, UNITE (Nilsson et al., 2019), and MycoBank (Robert et al., 2013) using a >95% sequence similarity cutoff.
Statistical analysis
To quantify alpha-diversity, we used Chao1 and Shannon index. To assess beta-diversity, we used Bray-Curtis dissimilarity and performed ordination analyses with non-metric multidimensional scaling (NMDS). Analysis of Similarity (ANOSIM) and permutational multivariate analysis of variance (PERMANOVA) tests were applied to test for significant differences in community composition. Analyses were performed after clustering at the family or genus level and using R version 4.4.1, and the phyloseq package (McMurdie and Holmes, 2013). Venn diagrams were generated using complete sample sets for each population and based on the top ten families for each population (https://bioinformatics.psb.ugent.be/webtools/Venn/).
Results
Comparison of bacterial microbiomes of D. suzukii across native and invasive ranges and lab-maintained populations
To perform a multi-region comparison of D. suzukii microbiomes, we analyzed the bacterial communities of samples collected from China, Japan, North America, Europe, the UK and Hawaiʻi. In terms of alpha-diversity, populations from the native range tended to have higher compositional richness and evenness compared to Europe, UK, and Hawaiʻi when grouped by family (Figure 1A, Table 1). In addition, almost all populations of D. suzukii exhibit distinct compositional profiles (ANOSIM p = 0.001, R = 0.61; Figure 1B, Table 2). However, one exception to this general pattern is that D. suzukii from the native ranges of China and Japan and the non-native range of Europe exhibited similar compositions (PERMANOVA p = 0.075; Table 2). Flies collected in Hawaiʻi, regardless of species, clustered together in the NMDS ordination plot (Figure 1B) although D. suzukii from Hawaiʻi differed significantly in compositional profile from other Hawaiian populations (PERMANOVA, p = 0.001; Table 2). Only two bacterial families were unique to flies collected in their native range of China and Japan (Flavobacteriaceae and Weeksellaceae), suggesting that this cosmopolitan species associates with microbes beyond those that are specific to its region of origin. Seven bacterial families were common to all wild-caught D. suzukii, potentially serving as a core microbial community: Acetobacteraceae, Anaplasmatacea, Erwiniaceae, Halomonadaceae, Pseudomonadacae, Sphingobacteriaceae, and Yersiniaceae (Figures 1C, D). At the genus level, only the endosymbiont Wolbachia was common to lab and wild flies (Supplementary Figure S2, Supplementary Table S6).
The bacterial community richness of lab populations was significantly lower compared to each of the wild populations, consistent with previous studies (Chandler et al., 2011; Staubach et al., 2013) (Figure 1A, Table 1). Lab flies from US, China, and Italy contained distinct communities of bacteria, with each profile correlating with geographical location (ANOSIM p = 0.001 R = 0.78; Supplementary Figure S1, Supplementary Table S7). However, there were only modest differences in terms of alphadiversity (Supplementary Table S7).
Bacterial and fungal gut microbiome comparison of native Hawaiian Drosophila, D. immigrans in Hawaiʻi, and D. suzukii in Hawaiʻi
We predicted that D. suzukii colonization of new ecological niches may rely on the incorporation of local microbes. To test this possibility, we compared the bacterial and fungal profiles of D. suzukii to another invasive species established in Hawaiʻi, D. immigrans, as well as to native Hawaiian Drosophila found at the same sites. In terms of community richness (Chao1), both invasive species exhibited significantly lower bacterial alpha-diversity compared to native flies (Figure 2A, Table 3). However, for fungal profiles, no significant differences in alpha-diversity were found between the three populations (Figure 2D, Table 3). Additionally, D. suzukii contained distinct bacterial communities compared to either D. immigrans or native Hawaiian flies at both family (ANOSIM p = 0.003, R = 0.18; Figure 2G) and genus levels (ANOSIM p = 0.012, R = 0.15; Supplementary Figure S2, Supplementary Table S6). Five families were found in D. suzukii that were not common to other Hawaiian populations. Of these, Rhizobiaceae was also not detected in D. suzukii from other regions and may reflect enrichment from the local environment. At the genus level, Citrobacter and Zymobacter appear to be enriched only in D. suzukii found in Hawaiʻi and no other Hawaiian populations or regions (Supplementary Figure S2). Four bacterial families were common to invasive and native species in Hawaiʻi whereas a single family is found in both invasive species, Moraxellaceae (Figure 2G). The presence of the Moraxellaxeae family in both Hawaiʻi D. immigrans and D. suzukii as well as D. suzukii populations outside of its native range may indicate a conserved ecological association with invasive drosophilids.
With respect to the fungal microbiome, native Hawaiian flies and both invasive populations exhibited similar profiles in terms of taxonomic composition (ANOSIM p = 0.01, R = 0.117) and alpha-diversity (Figure 2). Native and invasive species share five fungal families. As with bacteria, three fungal families appear unique to D. suzukii: Cordycipitaceae, likely a Diptera pathogen (Naranjo-Lázaro et al., 2014), Bulleribasidiaceae, which in its yeast state is used by D. suzukii as a food source (Jiménez-Padilla et al., 2020), and Chrysozymaceae, a yeast previously identified from ghost moth gut (Thitarodes sp.) (Liu et al., 2021).
Discussion
Microbes sourced from the local environment play myriad roles in host physiology including nutrient scavenging (Yamada et al., 2015; Bing et al., 2018), toxin inactivation (Kohl et al., 2014; Zhang et al., 2024), stress resilience (Houwenhuyse et al., 2021; Tefit et al., 2023; Price et al., 2025), lipid metabolism, and sleep regulation (Tefit et al., 2023). Given the broad range and global invasion of D. suzukii, we hypothesized that local microbial associations might accompany colonization and regional establishment. Specifically, we predicted that microbiomes of D. suzukii from different sites would contain different communities and more closely resemble those of local drosophilids. Alternatively, successful colonization could be aided by a core microbiome that is maintained regardless of host location. Our analysis of D. suzukii microbiomes collected from three continents and two islands provides evidence for both of these scenarios. While these results identify compositional trends rather than functional roles, they establish a valuable foundation for future experiments to test how microbiome variation influences colonization and host physiology.
Multi-region trends and local influences on the D. suzukii bacterial microbiome
Wild populations from each of the major sites exhibited distinct compositional differences, indicating geographically structured variation in microbiome composition. However, three of the seven families present in all wild D. suzukii, Acetobacteraceae, Erwiniaceae, and Pseudomonadaceae, were also found in a recent survey of wild D. suzukii populations from Oregon (USA) and Missouri (USA) (Bhandari et al., 2025), consistent with the possibility that taxa from these families form stable, mutualistic associations with D. suzukii. Two of the D. suzukii-associated bacterial families have species members with sugar production and cellulose degradation activities, and may facilitate D. suzukii’s ability to use multiple host fruits (Gao et al., 2023; Netrusov et al., 2023; Wünsche and Schmid, 2023; López-Hernández et al., 2025). Both Acetobacteraceae and Lactobacillaceae are considered facultative members of the D. melanogaster microbiome that promote host growth and participate in nutritional mutualism (Storelli et al., 2011; Pais et al., 2018). The Erwiniaceae family is also found in another widespread invasive insect, ambrosia beetles, for which it provides nutritional support and antibiotic protection (Cambronero-Heinrichs et al., 2023). The incorporation of bacterial taxa that offer enhanced metabolic capabilities is a common strategy observed in multiple invasive host species. Understanding the functional contributions of these microbes to D. suzukii adaptation may identify novel strategies for population control (Hamby and Becher, 2016).
Comparing the microbiomes of invasive species to native Hawaiian species
Although D. suzukii in Hawaiʻi share features of their microbial profile with populations from other regions, their microbiome composition more closely resembles that of other Hawaiian drosophilids. This outcome indicates that D. suzukii populations may both retain a subset of widespread bacterial families regardless of area of occurrence, yet enrich for other groups of local bacteria when colonizing a new habitat. For example, the Rhizobiaceae family, which appears to be enriched only in Hawaiʻi D. suzukii, belong to the Bacteroidetes phyla, members of which are capable of degrading simple and complex polysaccharidess (Vera-Ponce de León et al., 2020; Nweze et al., 2024). At the level of genus, two taxa appear to be enriched in Hawaiʻi D. suzukii: Zymobacter and Citrobacte, both of which are known to confer beneficial properties to host insects. Zymobacter facilitates sugar fermentation and is relatively abundant in field-collected mosquitoes and stingless bees (Hegde et al., 2018; Hall et al., 2020). In addition, Citrobacter provides resistances to insecticide and enhances development in black soldier flies (Cheng et al., 2017; Luo et al., 2023). Incorporation of microbes that potentially enhance metabolic capabilities may allow D. suzukii to expand its ecological niche. These associations may reflect conserved adaptation strategies in addition to widespread and consistent opportunistic colonization. Future studies of native vs. invasive flies from other sites are needed to determine whether this is a general strategy. Additionally, measurements of host range and adaptation paired with experimental manipulation of the microbiome will directly address the hypothesis that local microbe incorporation is a necessary step for successful colonization.
D. immigrans is considered a member of the guild of cosmopolitan Drosophila species, found worldwide (Nunney, 1996). Notably, the microbiome of D. immigrans, first reported in Hawaiʻi in 1948 (Mainland, 1949), more closely resembles that of endemic Hawaiian flies than D. suzukii, a species that arrived only in 1980 (Hauser, 2011). The extended establishment time of D. immigrans in Hawaiʻi compared to D. suzukii may have allowed the former to assimilate more of the local microbes. The mycobiome communities of invasive vs. native flies in the Hawaiian Island also exhibit similar compositional profiles. Given that flies obtain much of their microbiome through diet and environment interactions rather than vertical transmission (Douglas, 2019), the general overlap in fungal composition may reflect shared habitats among the native and invasive drosophilids in Hawaiʻi (Kaneshiro, 1983; Magnacca et al., 2008; Leblanc et al., 2009).
Overall, our findings reveal both geographic differentiation and partial overlap in D. suzukii microbiomes across continents and the Hawaiian Islands. These patterns are consistent with a core microbiome retained across regions and with enrichment of locally prevalent microbes. These results also suggest that plasticity in the D. suzukii microbiome may facilitate this species’ rapid colonization of novel ecological niches. Similar to a previous study of invasive Siganus fish in the Mediterranean Sea (Escalas et al., 2022), we identified a shift in microbiome composition when comparing D. suzukii from the native range in Asia to invaded ranges. Whether compositional change provides adaptive advantages (e.g., expanding dietary resources) remains an open question. Further functional exploration of how microbes contribute to host physiology and local adaptation through, for example, common garden transplant experiments with native vs. invasive hosts may lead to the development of new microbiome methods for population control. Incorporating microbiome manipulations and metagenomic analyses into invasion ecology could also clarify whether microbes act as opportunistic colonizers or facilitators of ecological expansion.
Supplementary Material
Supplemental Materials
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2025.1696606/full#supplementary-material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adrion JR, Kousathanas A, Pascual M, Burrack HJ, Haddad NM, Bergland AO, (2014). Drosophila suzukii: the genetic footprint of a recent, worldwide invasion. Mol. Biol. Evol 31, 3148–3163. doi: 10.1093/molbev/msu 24625158796 PMC 4245814 · doi ↗ · pubmed ↗
- 2Andreazza F, Bernardi D, dos Santos RSS, Garcia FRM, Oliveira EE, Botton M, (2017). Drosophila suzukii in Southern Neotropical region: current status and future perspectives. Neotropical Entomology 46, 591–605. doi: 10.1007/s 13744-017-0554-728852987 · doi ↗ · pubmed ↗
- 3Arisdakessian C, Cleveland SB, and Belcaid M (2020). “Meta Flow∣mics: Scalable and reproducible nextflow pipelines for the analysis of microbiome marker data,” in Practice and Experience in Advanced Research Computing (PEARC ‘20). (New York, NY, USA: Association for Computing Machinery).
- 4Bhandari R, Wong A-N, Lee JC, Boyd A, Shelby K, Ringbauer J Jr., (2025). Microbiome composition and co-occurrence dynamics in wild Drosophila suzukii are influenced by host crop, fly sex, and sampling location. Microbiol. Spectr 13, e 0260824. doi: 10.1128/spectrum.02608-2440793771 PMC 12403722 · doi ↗ · pubmed ↗
- 5Bing X, Gerlach J, Loeb G, and Buchon N (2018). Nutrient-dependent impact of microbes on Drosophila suzukii development. m Bio 9, e 02199–17. doi: 10.1128/m Bio.02199-1729559576 PMC 5874910 · doi ↗ · pubmed ↗
- 6Bolda MP, Goodhue R, and Zalom F (2010). Spotted wing Drosophila: potential economic impact of a newly established pest. Giannini Foundation Agric. Econ 13, 5–8.
- 7Calabria G, Máca J, Bächli G, Serra L, and Pascual M (2012). First records of the potential pest species Drosophila suzukii (Diptera: Drosophilidae) in Europe. J. Appl. Entomology 136, 139–147. doi: 10.1111/j.1439-0418.2010.01583.x · doi ↗
- 8Callahan BJ, Mc Murdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA 2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.386927214047 PMC 4927377 · doi ↗ · pubmed ↗