Shaping death: how the microbiome regulates tumour cell demise and therapy response
Martina Raudenska, Jan Balvan, Eliska Zgarbova, David Kalfert, Jan Plzak, Michal Masarik

TL;DR
This paper explores how the microbiome influences cancer cell death and therapy response, highlighting a bidirectional relationship with potential for precision cancer treatments.
Contribution
The paper provides a comprehensive review of the interplay between the microbiome and cancer cell death pathways, emphasizing key metabolic mediators.
Findings
The microbiome modulates cancer cell death pathways and therapy responses through microbial metabolites.
Cell death reshapes the microbiome by altering nutrients and immune signals.
Understanding this crosstalk could lead to microbiome-informed cancer therapies.
Abstract
Cell death is a fundamental process that maintains tissue homeostasis and shapes the tumour microenvironment. Cancer cells often evade or reprogram cell death pathways, which leads to malignancy and therapy resistance. On the other hand, the cell death of non-malignant cells significantly influences the adverse effects of anticancer therapy, such as the onset of mucositis or enteritis. Emerging evidence identifies both the tumour and gut microbiomes as critical modulators of these processes. In turn, cell death reshapes the microbial ecosystem by altering nutrient landscapes and immune signalling. Although both cell death and the microbiome are well-studied in cancer, their intersection remains underexplored. This review article addresses this gap by summarising how microbes and microbial metabolites modulate cancer cell death pathways and shape responses to anticancer therapy. We…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
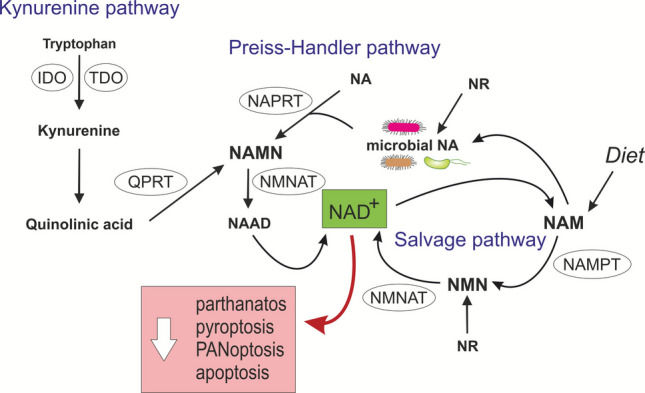 Figure 1
Figure 1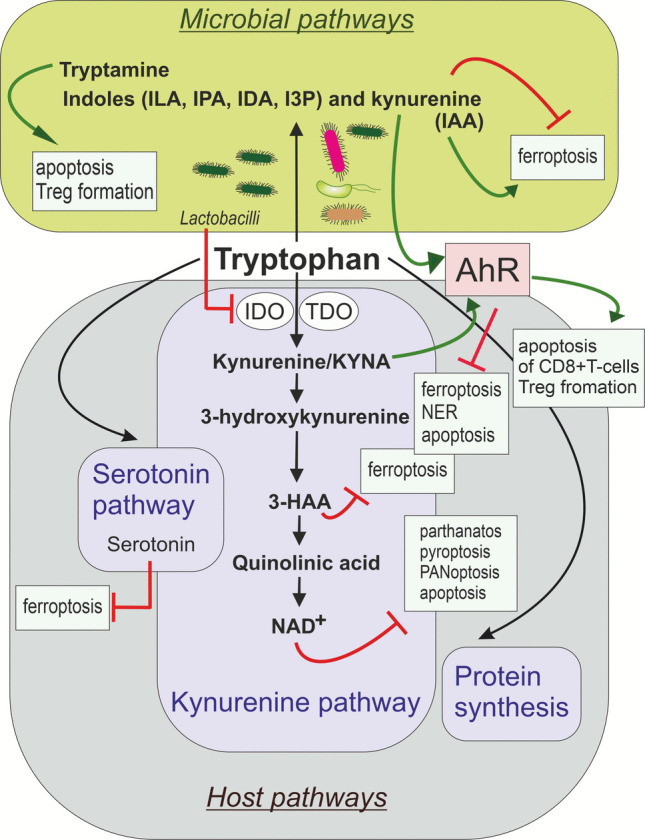 Figure 2
Figure 2 Figure 3
Figure 3- —Masaryk University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer Research and Treatments · Cancer, Hypoxia, and Metabolism · Gut microbiota and health
Introduction
Cell death serves as a crucial mechanism for maintaining physiological homeostasis by eliminating damaged or unnecessary cells, but it can also arise as a response to harmful stimuli. In cancer, the ability of cancer cells to evade or reprogram cell death pathways is central to malignancy [1]. Cell death is a fundamental process that further shapes the tumour microenvironment (TME), influencing tumour growth, immune activation, and therapeutic response [2, 3]. Recent studies have revealed that, beyond genetic and stromal determinants, the microbiome also plays a role in modulating cell death pathways. Microbial metabolites can directly or indirectly regulate cell death pathways in both cancer and TME cells. Anticancer therapies add further complexity as they trigger cell death while also altering the composition of the microbiome, microbial metabolism, and the favourable or unfavourable conditions for the growth of different bacterial species. For example, radiation therapy can alter short-chain fatty acid (SCFA)-producing bacteria, leading to changes in SCFA levels that may negatively impact cancer treatment outcomes [4]. Specifically, a reduced ratio of Bacteroidetes to Firmicutes after radiotherapy significantly affects the lipid profile of the intestinal epithelial barrier, leading to death of intestinal cells and progression of enteritis [5]. Chemoradiotherapy induces death of intestinal epithelial cells through various pathways, including apoptosis, necroptosis, and pyroptosis, leading to compromised gut barrier integrity and inflammation [6, 7]. Surprisingly, a recent study has shown that intestinal low-dose irradiation (ILDR, 1–3 Gy) induces an abscopal effect via distal immunostimulation, primarily through microbiota-driven alterations in systemic metabolism. ILDR modulates cholesterol and bile acid pathways, thereby enhancing the antigen-presenting capacity of dendritic cells (DCs). Elevated cholesterol accumulation in DCs further promotes antigen presentation and CD8⁺ T-cell activation without exhaustion. Certain Christensenella minuta strains further boost ILDR and PD-L1 blockade by promoting migration of intestinal dendritic cells to tumour-draining lymph nodes. A phase II trial confirms that ILDR can overcome immunotherapy resistance [8].
Microbiome-derived signals can modulate tumour cell death pathways, changing cancer cells' sensitivity to therapy, while tumour cells and their death reshape the local microbial community by changing nutrient and immune landscapes. Dying mammalian cells produce and shed metabolites that bacteria, such as Salmonella and E. coli, use as fuel to assist them in colonisation during inflammatory conditions or chemotherapy-induced mucositis [9]. Lung adenocarcinoma cells also create selective pressure favouring bacteria capable of synthesising L-methionine, an essential amino acid they cannot produce themselves. This mutualistic interaction enables bacteria to supply methionine to nutrient-deprived tumours and stop them from dying by ferroptosis [10], while cancer cells, in turn, promote the growth of methionine-producing bacteria [11]. Deciphering the bidirectional interplay between the microbiome and cell death regulation may provide a new therapeutic angle: leveraging microbiome modulation to promote immunogenic cell death pathways in tumours, while limiting resistance mechanisms. Such insights could guide the development of microbiome-based adjuvants to enhance standard therapies. Although cell death and microbiome are extensively studied in the context of cancer, the intersection between the two remains insufficiently explored. This review article aims to bridge the critical knowledge gap by providing a comprehensive overview of the current understanding of how microbial metabolites interact with cancer cell death pathways and influence responses to anticancer therapies.
Basic overview of regulated cell death pathways
The various forms of regulated cell death can be categorised into three distinct groups: suicide, sabotage, and murder. Suicide refers to forms of cell death that occur via molecular pathways evolved in response to signals. While these signals are not necessarily destructive in themselves, under certain circumstances, they can lead to cell death. Sabotage involves cell death that occurs when an active molecular process necessary for normal cell function becomes nonfunctional. Such forms of sabotage are autophagy-related cell death, cuproptosis, parthanatos, or ferroptosis. Murder involves cell death caused by another cell whose task is to kill the target cell. Within this broad framework, cells that die by apoptosis, pyroptosis, or necroptosis are said to commit suicide. Those that die by cytotoxic lymphocytes, antibody-dependent cytotoxicity, or complement-mediated lysis are murdered [12] (see Table 1). Table 1. Regulated Cell Death Categories (Based on Green [12])GroupDefinitionCell Death TypeShort DescriptionSuicideCells activate internally programmed death in response to internal or external signals that are not directly destructive but can trigger cell death pathwaysApoptosisApoptosis occurs via the intrinsic pathway (triggered by internal stress through mitochondria) or the extrinsic pathway (triggered by external death‑receptor signals); usually involves caspases, DNA fragmentation, and cell shrinkagePyroptosisInflammatory cell death triggered by inflammasomes; involves gasdermin pores and cell swelling/lysisNecroptosisRegulated necrotic death mediated by RIPK1–RIPK3–MLKL; results in membrane rupture and inflammationNETosisRegulated form of cell death in neutrophils. Neutrophils expel chromatin and granular proteins to form neutrophil extracellular traps (NETs) that immobilise and kill microbes; involves unique chromatin decondensation and lytic ruptureSabotageCell death occurs because a normally essential process becomes dysfunctional, leading to the collapse of homeostasisAutophagy-related cell deathExcessive autophagy leads to self-digestion and loss of essential cellular componentsCuproptosisCopper-induced cell death driven by disruption of mitochondrial metabolism and protein aggregationParthanatosPARP1-dependent death caused by massive DNA damage and NAD⁺/ATP depletion; AIF translocation to the nucleusFerroptosisIron-dependent lipid peroxidation overwhelms antioxidant defence, causing membrane damageMurderAn external cell or system kills the target cellCytotoxic lymphocyte–mediated killingKiller T-cells or NK-cells induce death via perforin–granzymes or death receptorsAntibody-dependent cellular cytotoxicityAntibodies bind the target; immune effector cells recognise Fc-fragments and induce cell deathComplement-mediated lysisComplement activation forms a membrane attack complex that punches holes in the target cell membrane
Cell suicide
The best-known form of cell suicide is apoptosis. Apoptosis is usually driven by caspase activation (however, caspase-independent apoptosis was also observed). Apoptosis occurs via two main routes: the intrinsic pathway, controlled by mitochondrial outer membrane permeabilization (MOMP), regulated by Bcl-2 family proteins, and the extrinsic pathway, triggered by death receptors [13]. Apoptotic cells undergo nuclear condensation, DNA fragmentation, and membrane blebbing, culminating in the formation of apoptotic bodies that are usually cleared by efferocytosis. The intrinsic apoptotic pathway is triggered by stress signals within the cell, such as DNA damage, oxidative stress, or growth factor deprivation. These signals activate pro-apoptotic proteins (e.g., BAX and BAK) from the Bcl-2 family, causing MOMP. This releases cytochrome c into the cytosol, where it forms an apoptosome complex with APAF-1 and pro-caspase-9. This leads to the activation of caspase-9 and subsequent executioner caspases, such as caspase-3; consequently, the cleaved Poly ADP-ribose polymerase (PARP) is formed. In the absence of anti-apoptotic proteins, activators such as BID, BIM, and PUMA can directly bind to and activate BAX and BAK. Additionally, sensitizers such as BAD, NOXA, BIK, and PUMA can suppress the inhibitory effect of BCL-2, BCL-XL, and MCL-1 on BAX and BAK, thereby indirectly activating them [14].
The extrinsic pathway of apoptosis is initiated by extrinsic ligands (e.g., FasL, TNF-α, and TRAIL) binding to death receptors (e.g., Fas/CD95 and TNFR1) on the cell surface. This leads to the formation of a death-inducing signalling complex (DISC), which activates the initiator caspase-8. Caspase-8 then activates the executor caspases, and the cleaved PARP is formed [14]. The extrinsic apoptotic pathway, which is controlled by death receptors, may proceed differently in different cell types. In adult lymphocytes or thymocytes (lymphocytes that originate in the thymus), i.e., cells that we refer to as cell type I, the proteolytic maturation of executor caspases, which is mediated by high levels of activated caspase-8, is usually sufficient to activate cell death. Therefore, apoptosis occurs without the involvement of mitochondria. In these cells, apoptosis cannot be inhibited by increased expression of anti-apoptotic Bcl-2 family proteins, or by the simultaneous deletion of BAX and BAK1, or the loss of BID [15, 16]. In type II cells (e.g., hepatocytes, pancreatic β-cells, and most tumour cells), the external signal alone is insufficient to activate enough caspase-8 to trigger apoptosis. The activation of caspases-3 and −7 is prevented in type II cells by the XIAP protein [17], and thus the extrinsic apoptotic pathway also requires the proteolytic cleavage of BID for successful activation [18]. The cleaved form of BID (tBID), which is produced in this process, is transported to the outer mitochondrial membrane. There, it acts on BAX/BAK-dependent mitochondrial outer membrane permeabilisation (MOMP) and subsequent apoptosome assembly, which is accompanied by caspase-9 activation. While basal XIAP levels are similar in type I (thymocyte) and type II (hepatocyte) cells, stimulation of the FAS receptor leads to a rapid decrease in XIAP levels in thymocytes, but an increase in hepatocytes [17, 19]. Therefore, it can be concluded that type I cells undergo apoptosis independently of mitochondria, whereas type II cells undergo MOMP.
Necroptosis is primarily initiated via death receptors, such as TNFR1. The most widely studied form to date is TNF-α-induced necroptosis, but many other stimuli capable of activating necroptosis have been identified. These include CD95L (also known as FASL), TRAIL, TWEAK, genotoxic stress, polyclonal T-cell receptor stimulation, viral activation of Z-DNA binding protein 1 (ZBP1, also known as DAI), and certain anticancer drugs, such as shikonin and GX15-070 [20]. During necroptosis, RIPK1 becomes activated and interacts with RIPK3 to form a multiprotein complex known as the necrosome. Within the necrosome, RIPK3 phosphorylates the pseudokinase MLKL, which then forms oligomers and moves to the plasma membrane. There, phosphorylated MLKL disrupts the membrane's integrity, leading to cell swelling and ultimately lysis. Necroptotic cells release damage-associated molecular patterns (DAMPs), stimulating immune responses. The interaction of active MLKL with the plasma membrane requires certain inositol phosphate forms generated by lipid kinases (e.g., IP5 and IP6); without these, necroptosis either does not occur or is reduced [12].
Pyroptosis is mostly triggered by intracellular pattern recognition receptors (PRRs) detecting pathogenic microorganisms or dangerous signals. This leads to the formation of a multiprotein complex called an inflammasome, which activates caspase-1. The best-characterised proteins in inflammasomes are NLRP1, NLRP3, NLRC4, and AIM2 [21]. Activated caspase-1 then cleaves gasdermin D (GSDMD). The N-terminal fragment of GSDMD then inserts itself into the plasma membrane, forming pores. GSDMD pores favour ninjurin 1 (NINJ1)-induced plasma membrane rupture and cell death [22]. This leads to cell swelling, lysis, and the release of inflammatory interleukins (IL-1β and IL-18), amplifying immune signalling. In some contexts, pyroptosis can also be initiated by caspase-4 or −5 (in humans) or caspase-11 (in mice) in response to intracellular lipopolysaccharide (LPS) during infection with gram-negative bacteria, leading to non-canonical inflammasome activation [23]. Under certain circumstances, the N-terminal domain of GSDMD can be translocated to the outer mitochondrial membrane, causing permeabilisation independently of BAX and BAK [24]. Damaged mitochondria may further facilitate the formation of NLRP3 inflammasomes, as destabilised mitochondria induce cardiolipin externalisation, which promotes NLRP3 inflammasome assembly and pyroptosis initiation [25].
NETosis is a regulated form of cell death in neutrophils that helps defend the host against pathogens. It is initiated when neutrophils are activated by microbes, proinflammatory cytokines, or chemical stimuli. This triggers a cascade involving reactive oxygen species (ROS), which drive chromatin decondensation. Neutrophil elastase (NE) translocates to the nucleus and cleaves histones, while myeloperoxidase (MPO) aids chromatin relaxation and nuclear envelope breakdown. The nuclear and granular contents then mix, and the plasma membrane ruptures, releasing neutrophil extracellular traps (NETs) composed of chromatin and histones. These NETs capture and kill pathogens, limiting their spread. Many bacteria trigger NET formation, or NETosis, including species like Staphylococcus aureus, Klebsiella pneumoniae, Fusobacterium nucleatum, and Mycobacterium bovis [26]. The mechanism can involve direct detection of bacteria or recognition of bacterial byproducts, including lactate, which signals the release of NETs [27]. NETs protect cancer cells by forming a physical barrier around them, shielding tumour cells from cytotoxic T-lymphocytes and NK-cells by masking their interaction sites. Additionally, NETs promote metastasis by enhancing tumour cell migration and invasion [28]. Nuclease expression by Staphylococcus aureus facilitates degradation of NETs [29].
Cell sabotage
Many of the newly discovered forms of cell death can be categorised as sabotage. While these are active forms of cell death, they did not evolve primarily for the purpose of killing cells. One example of a "sabotage" type of cell death is ferroptosis, which occurs when the mechanisms necessary to prevent or detoxify lipid peroxides in membranes fail. Unlike apoptosis or necroptosis, ferroptosis does not involve the activation of caspases. Instead, it is driven by the accumulation of ROS, which specifically oxidise polyunsaturated fatty acids (PUFAs) in cell membranes. Lipid peroxidation of the PUFAs occurs via iron-dependent Fenton reactions or enzymatic pathways involving lipoxygenases, cyclooxygenases, and cytochrome P450, producing hydroperoxides that amplify ferroptotic signalling. Enzymes such as acyl-CoA synthetase long chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) incorporate PUFAs into membrane phospholipids, thereby increasing cellular susceptibility to ferroptosis. Several key mechanisms, including iron metabolism, the mode of lipid peroxidation, and antioxidant defence, tightly regulate this process. The enzyme glutathione peroxidase 4 (GPX4) plays a central protective role, reducing lipid hydroperoxides to non-toxic lipid alcohols with the help of glutathione (GSH) [30]. Numerous studies have suggested that inducing ferroptosis could affect the effectiveness of tumour treatment and even reverse treatment resistance [31]. The levels of intracellular iron and iron uptake are significantly higher in rapidly proliferating cells, such as tumour cells. Furthermore, tumour cells often exhibit abnormal lipid metabolism and increased ROS production. These factors sensitise malignant cells to ferroptosis. However, cancer cells counter this problem by increasing the activity of ferroptosis-inhibiting mechanisms, such as GPX4. Therefore, ferroptosis inducers, including GPX4 inhibitors, are somewhat specific in their targeting of cancer cells [32]. Some chemotherapeutic agents used to treat solid tumours are likely to trigger ferroptosis. For instance, altretamine, a DNA-alkylating agent commonly employed in the treatment of late-stage ovarian cancer, induces ferroptosis by inhibiting GPX4. Recently, sulfasalazine, a non-steroidal anti-inflammatory drug traditionally used to treat rheumatoid arthritis, has been shown to induce ferroptosis in head and neck tumours. Sorafenib, a multikinase inhibitor (VEGFR, PDGFR, c-Kit and RET) used to treat hepatocellular carcinoma, can also induce ferroptosis [33].
Microbes can influence ferroptosis by altering host iron metabolism. Many bacteria found in the TME secrete siderophores that scavenge extracellular iron, depriving host cells of iron needed for lipid peroxidation and thereby limiting ferroptosis. These bacteria include Pseudomonas aeruginosa, Escherichia coli, and Staphylococcus spp., which produce siderophores like pyoverdine (from P. aeruginosa), enterobactin (from E. coli), and staphyloferrin A (from Staphylococcus spp.) to acquire essential iron for their survival and growth within the tumour [34, 35]. Probiotics, such as Lactobacillus plantarum, can inhibit ferroptosis by releasing specific extracellular vesicles [36] and reducing oxidative stress [37]. Some studies show that gut microbiota–derived metabolites can regulate ferroptosis via the GPX4/Nrf2 pathway. For example, 3-hydroxyphenylacetic acid and capsiate upregulate GPX4 to inhibit ferroptosis and protect against aging-related spermatogenic dysfunction or intestinal ischemia reperfusion injury [38, 39]. Conversely, dysbiosis with harmful bacteria like adherent-invasive E. coli reduces GPX4 and ferritin, promoting lipid peroxidation and ferroptosis in intestinal epithelial cells [40]. P. aeruginosa promotes ferroptosis by degrading the key anti-ferroptotic enzyme GPX4 via chaperone-mediated autophagy, while simultaneously increasing the labile iron pool and driving ROS-dependent lipid peroxidation [41]. Thus, microbial control of iron availability can either suppress or enhance ferroptotic cell death, shaping infection outcomes and potentially influencing cancer progression.
Another type of 'sabotage' cell death is parthanatos. This type of cell death is primarily caused by the excessive activation of an enzyme called poly(ADP-ribose) polymerase-1 (PARP-1). PARP-1 is normally activated in response to DNA damage to facilitate its repair, but excessive damage can lead to hyperactivation of the enzyme. This results in the depletion of cellular NAD⁺ and ATP, as well as the disruption of metabolic homeostasis. A distinctive feature of parthanatos is the accumulation of poly(ADP-ribose) (PAR) polymers, which are transported from the nucleus to the mitochondria, where they promote the release of apoptosis-inducing factor (AIF). Once released, AIF moves to the nucleus, where it induces extensive DNA fragmentation and chromatin condensation independently of caspases [42]. Bacteria can promote parthanatos through several interconnected mechanisms. Pathogens such as Helicobacter pylori and Escherichia coli produce genotoxins (e.g., CagA, VacA, colibactin) that induce DNA damage and drive PARP1 hyperactivation [43, 44]. In parallel, NADase (an enzyme that consumes NAD⁺) from Streptococcus pyogenes, directly depletes cellular NAD⁺ pools, sensitising host cells to PARP1-dependent energy collapse [45]. This mechanism can contribute to significant oncolytic (cancer-killing) activity of Streptococcus pyogenes, which was observed in various cancers, including glioma, hepatoma, and pancreatic cancer [46].
Cell death types classified as "sabotage" also include cuproptosis and necrosis controlled by mitochondrial permeability transition (MPT-controlled necrosis). Cuproptosis is a form of RCD caused by intracellular accumulation of copper ions and is closely linked to mitochondrial metabolism. It occurs when excess copper binds directly to lipoylated components of the Krebs cycle in mitochondria, leading to protein aggregation and proteotoxic stress. Cells with high mitochondrial activity and dependence on oxidative phosphorylation are particularly susceptible to cuproptosis. The discovery of cuproptosis has opened up new possibilities for potential therapy, particularly in tumours with high mitochondrial respiration, by exploiting their sensitivity to copper-induced toxicity [47]. MPT-mediated necrosis is controlled by a change in mitochondrial permeability and is initiated by severe oxidative stress and increased cytosolic Ca^2+^ concentration. A sudden loss of inner mitochondrial membrane permeability causes a collapse in mitochondrial membrane potential and inhibits ATP synthesis. It also triggers a profound ionic imbalance, which leads to the osmotic breakdown of organelles and cell death [48].
Autophagy between cytoprotection and cell demise
Autophagy is a highly conserved catabolic process that maintains cellular homeostasis by degrading and recycling cytoplasmic components via the lysosomal pathway. The ULK1 complex (comprising ULK1/2, ATG13, FIP200, and ATG101) is responsible for initiating its molecular mechanism, which is typically activated under conditions of nutrient deprivation or stress, when the energy sensor AMPK (AMP-activated protein kinase) is activated, and the growth regulator mTORC1 is inhibited. This triggers the recruitment of the class III PI3K complex I (Beclin 1, VPS34, VPS15, ATG14L), which generates phosphatidylinositol 3-phosphate (PI3P) at the nascent phagophore, allowing the recruitment of downstream ATG proteins. Expansion of the phagophore is driven by ubiquitin-like conjugation systems that coordinate the activity of ATG proteins together with LC3 (microtubule-associated protein 1 light chain 3) and GABARAP family members. The ATG12–ATG5–ATG16L1 complex enhances the final connection of phosphatidylethanolamine (PE) molecules, resulting in the formation of membrane-bound LC3-II and/or GABARAP-PE. This process drives membrane elongation and closure, producing the autophagosome. This double-membrane vesicle engulfs damaged organelles, protein aggregates, or surplus cytoplasm, and subsequently fuses with lysosomes, where lysosomal hydrolases degrade the contents. In addition to managing autophagy induction in complex I, VPS34-Beclin1 also has a role in the fusion of autophagosomes with lysosomes as complex II. Metabolites (amino acids, fatty acids, nucleotides) obtained by autophagy are recycled to sustain ATP production and biosynthesis, especially under stress conditions such as nutrient deprivation, hypoxia, infection, or therapeutic insult [49].
Microbes can modulate autophagy in cancer cells, influencing tumour progression and therapy resistance. For example, Porphyromonas gingivalis induces autophagy in oral cancer cells [50]. Fusobacterium nucleatum promotes chemoresistance in colorectal and oesophageal cancers by upregulating autophagy-related proteins (ULK1, ATG7, LC3, Beclin1) and enhancing epithelial–mesenchymal transition (EMT) [51–53]. Knockdown of ATG7 reverses this resistance, highlighting its key role [52]. Salmonella typhimurium strains (A1-R, VNP20009) trigger autophagy as a host defence mechanism. Blocking autophagy (via ATG5/Beclin1 knockdown or autophagy inhibitors, such as chloroquine and bafilomycin A1) enhances bacterial-mediated cancer cell killing [54]. Meanwhile, cytolethal distending toxin (CDT) produced by Campylobacter jejuni suppresses irradiation-induced autophagy by reducing acidic vesicular organelle formation, increasing radiosensitivity in prostate cancer [55].
Mitophagy is a selective form of autophagy that ensures mitochondrial quality control by removing damaged or superfluous mitochondria, thereby maintaining cellular homeostasis and metabolic balance. The process is initiated when dysfunctional mitochondria lose their membrane potential, a signal that recruits key regulatory proteins to the outer mitochondrial membrane. The best-characterised pathway involves the serine/threonine kinase PINK1 (PTEN-induced kinase 1) and the E3 ubiquitin ligase Parkin. Under normal conditions, PINK1 is imported into healthy mitochondria and rapidly degraded. However, upon mitochondrial depolarisation, PINK1 accumulates on the outer mitochondrial membrane, where it phosphorylates ubiquitin and Parkin, leading to Parkin activation. Activated Parkin ubiquitinates multiple outer mitochondrial membrane proteins, creating a signal for the recruitment of autophagy adaptors such as p62/SQSTM1, NBR1, OPTN, and NDP52, which bridge ubiquitinated mitochondria with the autophagy machinery through their LC3-interacting regions. In parallel, Parkin-independent pathways also exist, involving receptors such as BNIP3, NIX, and FUNDC1, which reside on the mitochondrial membrane and directly interact with LC3 via LC3-interacting regions (LIR motifs), bypassing ubiquitination. Ultimately, the isolation membrane, or phagophore, engulfs the damaged mitochondrion, forming a mitophagosome that fuses with lysosomes for degradation. Through these mechanisms, mitophagy plays a dual role: under physiological conditions, it preserves mitochondrial function and prevents oxidative stress, while under excessive or dysregulated activation, it may contribute to cell death [56].
Although autophagy is predominantly a pro-survival mechanism, preventing toxic accumulation and maintaining metabolic balance, it exists at the boundary between survival and death. Excessive or dysregulated autophagy can result in autophagy-related cell death, a regulated form of cell death distinct from apoptosis and necrosis, where uncontrolled self-digestion compromises essential cellular structures. Autophagy also exhibits extensive crosstalk with other death pathways: for example, it can inhibit apoptosis by degrading damaged mitochondria and reducing cytochrome c release, or conversely, promote ferroptosis by influencing iron homeostasis. Following ferritinophagy induction (ferritinophagy is a selective form of autophagy that specifically targets intracellular ferritin), autophagolysosomes degrade ferritin, resulting in the release of free Fe^2+^. This iron fuels the Fenton reaction, amplifying the production of ROS and ferroptosis [57].
Cell death at the crossroads
For many years, it was generally assumed that, once the process of cell death was activated, it could not be reversed, and the cell was doomed to die. However, it now appears that, despite the molecular activation of a certain type of cell death, some cells can survive this 'near-death experience', which can affect their properties and behaviour. While the extensive MOMP usually seals the fate of stressed cells by triggering the activation of caspases, the partial permeabilisation of the outer mitochondrial membrane (sublethal MOMP) has been associated with tumour cell survival, increased genomic instability, and tumour progression. Sublethal MOMP also triggers a stress adaptation pathway involving cytochrome c, EIF2AK1 and ATF4. This mechanism confers a resistant phenotype to tumour cells that survive sublethal apoptotic signalling, which is associated with increased metastatic spread [58]. The reversibility of certain cell death mechanisms represents a significant window of opportunity for bacterial effects. The microbial signals can shift the balance between survival and death, influencing tumour progression, immune activation, and therapeutic responses. For example, Chlamydia trachomatis inhibits apoptosis in infected cells by targeting the pro-apoptotic proteins (BAX, BAK) [59] and p53 degradation [60], creating the possibility of sublethal apoptotic signalling. Chlamydia trachomatis also alters the expression of ferroptosis-related proteins and inhibits ferroptosis via the p53-SLC7A11 axis [61].
Cell death is now understood not as a fixed choice between distinct pathways, but as a dynamic network, allowing cells to switch between death programmes depending on context. A key example is the interplay between apoptosis and necroptosis, both regulated by TNF receptors and sharing FADD and caspase-8. Normally, caspase-8 suppresses necroptosis by cleaving RIPK1 and RIPK3, favouring apoptosis. When caspase-8 is inhibited (e.g., by viral infection or mutation), necroptosis proceeds via the RIPK1–RIPK3–MLKL axis, ensuring cell elimination even if apoptosis fails [62]. A clinical example would be infection with Coxiella burnetii. C. burnetii inhibits TNFα-mediated caspase-8 activation, thereby blocking apoptosis and increasing susceptibility to necroptosis [63]. Coxiella burnetii has a demonstrated role in the tumour microenvironment, with its presence associated with an increased risk of B-cell non-Hodgkin lymphoma [64] and the formation of granulomas [65]. In some apoptosis-resistant cancer cells, ubiquitination of RIPK1 via cellular apoptosis inhibitors (cIAPs) or the LUBAC complex can activate NF-κB, promoting survival and inflammation [66]. Indeed, LUBAC determines chemotherapy resistance in squamous cell lung cancer [67]. Shigella flexneri suppresses NF-kB nuclear translocation in response to IL-1β and TNF by targeting the LUBAC component HOIP for proteasomal degradation [68]. Some studies showed that Shigella flexneri has the potential to induce pro-apoptotic effects in pancreatic cancer cells [69] or macrophages [70].
Pyroptosis and apoptosis are also interconnected. When apoptosis is blocked (e.g., caspase-8 inhibition), caspase-1 can trigger pyroptosis[71, 72]. In GSDMD-deficient cells, caspase-1 induces apoptosis via the BID/caspase-9/caspase-3 axis, sometimes followed by GSDME-mediated pyroptosis. Since GSDME is often silenced in tumours but not healthy tissues, caspase-3 activation may underlie chemotherapy-related toxicities [73, 74]. On the other hand, pyroptotic caspase-1 can cleave BID to activate apoptosis, particularly in cells with little or no GSDMD (e.g., neurons, mast cells). While inflammasomes induce pyroptosis, caspase-1 also activates caspase-3 and 7, which inactivate GSDMD, creating feedback to limit inflammation [14]. This mechanism is co-opted by Shigella dysenteriae (specifically serotype 1) and Shiga toxin-producing Escherichia coli. Shiga toxin recruits caspase-3 to cleave an active pore-forming N-terminal fragment of GSDMD, thereby preventing the formation of membrane pores required for pyroptosis [75].
PANoptosis is a recently proposed concept in cell death biology that describes a coordinated and interconnected form of cell death, integrating features of pyroptosis, apoptosis, and necroptosis. The term highlights that cell death is not always a linear or isolated process; instead, cells may activate multiple overlapping death programmes depending on the type and intensity of stress. PANoptosis involves simultaneous or sequential activation of apoptotic caspases (e.g., caspase-3, caspase-8), pyroptotic inflammasomes (e.g., caspase-1, gasdermins), and necroptotic mediators (RIPK1, RIPK3, MLKL). It can be induced by microbial infections, inflammatory signals (such as TNF-α or interferons), or genotoxic and metabolic stress [76]. ZBP1 (Z-DNA binding protein 1) is an innate immune receptor that senses nucleic acids in the unusual Z-conformation and triggers PANoptosis. Upon activation, ZBP1 forms a complex with RIPK3 and caspase-8, creating a signalling scaffold that drives NLRP3 inflammasome–dependent pyroptosis, caspase-8–mediated apoptosis, and RIPK3–MLKL necroptosis [77]. Z-DNA has been found in the matrix of Streptococcus mutans biofilms and in dental biofilms. Streptococcus mutans has been shown to promote the progression of oral squamous cell carcinoma (OSCC) through inflammation and NF-κB activation [78, 79]. Mechanistically, Streptococcus mutans infection can promote chronic oral inflammation through persistent activation of innate immune sensors (such as ZBP1). This can trigger PANoptosis and the release of pro-inflammatory cytokines and DAMPs, sustaining a tumour-promoting microenvironment. In OSCC, where apoptosis pathways are often dysregulated [80], PANoptosis-related inflammation may represent an underexplored driver of OSCC progression. Bacteria and LPS strongly enhance inflammatory cell death via the PANoptosis pathway, involving caspase-1, caspase-11, caspase-8, and RIPK3, with caspase-7 also contributing. Additionally, NINJ1 acts as a key executioner mediating the release of inflammatory molecules independently of gasdermin D, gasdermin E, and MLKL [81].
Some proteins and molecules, such as ROS, nicotinamide adenine dinucleotide (NAD⁺), and p53, occupy central positions within the network of interconnected cell death pathways. Modulating their activity can influence multiple forms of regulated cell death. For example, p53 induces apoptosis in response to DNA damage by activating pro-apoptotic genes such as PUMA, BAX, and NOXA. It also acts through transcription-independent mechanisms, binding anti-apoptotic proteins (BCL-2, BCL-XL, MCL-1) or directly activating BAK, thereby promoting intrinsic patway of apoptosis [82]. It can also promote ferroptosis by repressing the cystine/glutamate antiporter (SLC7A11), leading to lipid peroxidation [83, 84]. In addition, p53 contributes to necroptosis via downregulation of sulfiredoxin and peroxiredoxin [85] and can modulate autophagic flux by controlling genes like DRAM1 [86]. p53 also regulates the biogenesis of iron-sulphur clusters and the copper chelator glutathione, key components of the cuproptotic pathway, indicating a potential role for p53 in cuproptosis [87]. Importantly, the type of cell death induced often depends on cellular context, stress intensity, and crosstalk with other signalling pathways. Consequently, microbiome and microbial metabolites can shift the balance between survival and death or types of cell death. The distinct form of cell death not only determines cellular fate but also shapes tissue-level and systemic outcomes. This demonstrates the potential of the microbiome to not only influence tumour progression but also shape therapeutic responses by fine-tuning the network of cell death pathways. For example, gut microbes profoundly influence p53 signalling in colorectal cancer. Pathogenic E. coli strains, particularly pks + E. coli, contribute to tumorigenesis by producing colibactin, which induces a senescence-associated secretory phenotype [88] and increases p53 mutations [89], thereby promoting cancer progression through the Wnt/β-catenin pathway [90]. Colibactin synthesis by pks + E. coli requires spermidine, a polyamine abundant in cancer tissues and produced by gut microbes. Loss of spermidine synthase reduced colibactin-induced DNA damage, which was restored by spermidine supplementation [91, 92]. OmpA protein was overexpressed in E. coli from the B2 phylogenetic group isolated from colorectal cancer (CRC) patients, and it markedly suppressed the expression of the pro-apoptotic genes BAX and BAK in the HCT116 cells [93]. Conversely, probiotics such as Lactobacillus rhamnosus and L. plantarum enhance p53 and BAX expression, promoting apoptosis and suppressing tumour growth [94, 95]. Another gut probiotic, Akkermansia muciniphila, also upregulates p53 [96]. Together, these findings underscore the role of gut microbes in modulating cell death through p53 pathways.
NAD⁺– microbiome axis in cell death control
Nicotinamide adenine dinucleotide (NAD⁺) is a pivotal metabolite that connects energy metabolism, DNA repair, and redox balance. Due to its role in the cell's antioxidant defence, NAD⁺ availability directly influences several forms of regulated cell death, positioning it as a central node in determining cell fate under stress conditions [97, 98]. Apoptosis is closely linked to NAD⁺ homeostasis through the activity of the NAD⁺-dependent histone deacetylase SIRT1. By removing acetyl groups from lysine residues, SIRT1 regulates key apoptotic pathways [99]. For instance, deacetylation of p53 (e.g., at C-terminal lysine 382) [100] reduces its ability to activate pro-apoptotic genes such as PUMA, thereby suppressing p53-dependent apoptosis [99, 101]. SIRT1 also modulates BCL-2 family proteins [99], represses FOXO-driven apoptosis under stress [102], and attenuates NF-κB signalling to limit inflammasome activation [103]. Importantly, NAD⁺ precursors such as nicotinic acid and nicotinamide provide cytoprotection against apoptosis induced by oxidative and ER stress, further emphasising the role of NAD⁺ in maintaining cell survival under stress [104]. Beyond apoptosis, SIRTs also regulate autophagy by modulating Atg proteins or AMPK activation, which promotes autophagy and mitigates apoptosis in hypoxic stress [105]. SIRT1 transient upregulation is sufficient to enhance basal autophagy [106]. SIRT1 plays a central role in autophagy regulation by deacetylating Atg proteins (Atg5, Atg7, Atg8) [106] as well as Beclin-1, LC3, and ULK1, which together influence autophagic flux and autophagosome–lysosome fusion. Depending on the context, these actions can either promote or impair autophagy, with SIRT1 loss leading to autophagy dysregulation [99].
Severe DNA damage activates PARP-1, causing rapid depletion of NAD⁺ and ATP and accumulation of PAR, which disrupts glycolysis and energy balance, ultimately leading to cell death. Restoring NAD⁺ can reverse these effects as extracellular NAD⁺ enhances PARP-dependent DNA repair capacity [107]; for example, NAD⁺ supplementation protects mouse astrocytes from PARP-1–induced glycolytic inhibition and cell death [108]. Similarly, in models of septic shock, NAD⁺ administration has been shown to block pyroptosis and improve survival outcomes [109]. Moreover, NAD⁺ treatment prevents rotenone-induced apoptosis and necrosis in pheochromocytoma cells [110]. Under oxidative stress, NAD⁺ dynamics dictate cell death mode. In H_2_O_2_-treated HeLa cells, PARP1 depletes NAD⁺. Under mild stress, nicotinamide phosphoribosyl transferase-dependent salvage restores NAD⁺ and ATP synthesis, supporting ATP-dependent apoptosis. Severe stress impairs NAD⁺ recovery, blocking apoptosis and promoting necrosis, highlighting NAD⁺ metabolism as a key switch between cell death pathways [97]. Beyond individual pathways, NAD⁺ depletion is sensed by NLRC5, which assembles the PANoptosome to trigger PANoptosis while promoting inflammation [111]. These findings highlight NAD⁺ as a central metabolic hub that not only governs energy homeostasis but also orchestrates complex, integrated cell death and inflammatory responses, highlighting its broad role across distinct cell death pathways. NAD⁺ also plays a key role in antitumour immune responses, as regulatory T cells (Tregs; immunosuppressive immune cells that hinder anti-tumour responses and promote cancer progression) are susceptible to NAD⁺-induced cell death [112].
Except for neurons, mammalian cells cannot import NAD⁺ and must instead produce it. NAD⁺ can be produced via two main pathways: de novo synthesis and the salvage pathway (Fig. 1) [113]. De novo synthesis occurs through either the kynurenine pathway, which builds NAD⁺ from the amino acid tryptophan and the vitamin nicotinic acid (NA, niacin) via the kynurenine pathway with the help of enzymes tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO), or the Preiss-Handler pathway, which transforms NA to NAD⁺ via nicotinic acid phosphoribosyl transferase (NAPRT). The salvage pathway recycles nicotinamide, NA, nicotinamide riboside (NR), and nicotinamide mononucleotide (NAM) back to NAD⁺, with nicotinamide phosphoribosyl transferase (NAMPT) controlling the rate-limiting step [114]. NR supports NAD⁺ mainly via microbial conversion to NA, underscoring the microbiome’s role [115].Fig. 1. Nicotinamide adenine dinucleotide (NAD⁺) metabolism and its connection with cell death. NAD⁺ is a key metabolite linking energy metabolism, DNA repair, and redox homeostasis. By supporting the cell’s antioxidant defence, NAD⁺ availability critically regulates multiple forms of programmed cell death, making it a central determinant of cell fate under stress. NAD⁺ can be synthesised through three major biochemical routes: the kynurenine pathway, the Preiss–Handler pathway, and the salvage pathway. In the kynurenine pathway, kynurenine is produced from tryptophan, with Tryptophan 2,3-dioxygenase (TDO) and Indoleamine 2,3-dioxygenase (IDO) acting as rate-limiting enzymes. Kynurenine then undergoes several conversions to yield quinolinic acid. Quinolinic phosphoribosyltransferase (QPRT) converts quinolinic acid into nicotinic acid mononucleotide (NAMN), which enters the Preiss–Handler pathway. In the Preiss–Handler pathway, nicotinic acid (NA) is converted to NAMN by nicotinic acid phosphoribosyltransferase (NAPRT). NAMN is then converted to nicotinic acid adenine dinucleotide (NAAD) by nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1), and finally to NAD⁺ by NAD⁺ synthase (NADS). In the salvage pathway, NAD⁺-consuming reactions generate nicotinamide (NAM) as a by-product, which is recycled into NAD⁺. NAM is first converted to nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of this pathway, and NMN is subsequently converted to NAD⁺ by NMNAT1–3. Nicotinamide riboside (NR) can serve as an additional precursor that is phosphorylated by nicotinamide riboside kinase 1 or 2 (NRK1/2) to generate NMN, linking it to the salvage pathway. Host-derived nicotinamide (NAM) enters the gut to support microbial NAD⁺ synthesis, while microbes convert NAM into nicotinic acid (NA), which is absorbed by intestinal tissues and used for NAD⁺ regeneration via the Preiss–Handler pathway. Because NAPRT is not feedback-inhibited like NAMPT, NA supplementation efficiently boosts NAD⁺. Microbial conversion of NAM and NR helps sustain circulating NA and host metabolic flexibility, maintaining normal NA levels even in the absence of dietary precursors
Most tissues depend on the salvage pathway for NAD⁺ regeneration, but the Preiss-Handler pathway becomes more important under high NA availability. NAD⁺ precursors are produced both by the host and the gut microbiome, forming a symbiotic cycle in NAD⁺ metabolism. Host-derived nicotinamide (NAM) enters the gut to fuel microbial NAD⁺ synthesis, while microbes convert it into NA that can be taken up by host intestinal tissues and used to regenerate NAD^+^ via the Preiss-Handler pathway. Unlike NAMPT, NAPRT is not feedback-inhibited, making NA supplementation an efficient way to boost NAD⁺. The microbiome-driven transformation of precursors, including NAM and NR, is essential for maintaining circulating NA and supporting the host’s metabolic flexibility; even with a diet free of both NAM and NA, microbial NA production remains high enough to maintain normal levels of circulating NA in the host [115]. Moreover, NA (as well as butyrate [116]) activates the GPR109A receptor on epithelial and immune cells, where it can exert anti-inflammatory and tumour-suppressive effects, including cytokine regulation, immune cell differentiation, and induction of apoptosis. GPR109A is expressed in normal mammary tissue but silenced in breast tumours and cancer cell lines. Restoring its function reduces cAMP, induces apoptosis, and suppresses tumour growth, while GPR109A deletion in mice accelerates mammary tumorigenesis and metastasis [117].
NAD⁺ in the context of cancer
While ATP is essential for cancer cell function, some research suggests that the demand for NAD⁺ may be even greater than that of ATP in rapidly proliferating cells, as it is crucial for maintaining cellular redox balance to prevent cell death. NAD⁺ regeneration is also essential for de novo lipid synthesis. In the absence of exogenous lipids, limited NAD⁺ availability impaired cell proliferation and lipid synthesis, but supplementation with alternative electron acceptors (pyruvate, α-ketobutyrate, or NADH oxidase from Lactobacillus brevis) restored proliferation [118, 119]. Li et al. also showed that acetate (as a direct carbon source for cytosolic acetyl-CoA) can bypass the NAD⁺ consuming reactions involved in de novo lipid synthesis [119]. The increased NAD⁺ requirement may explain why cancer cells rely heavily on lactate fermentation. Lactate fermentation regenerates NAD⁺ by converting NADH (produced during glycolysis) back to NAD⁺ as pyruvate is converted to lactate (reaction catalysed by the enzyme lactate dehydrogenase). Normally, cells produce NAD⁺ and ATP together through respiration. However, if a cell has excess ATP, it can slow down the entire process, including NAD⁺ production. By increasing lactate production, a cell can bypass this bottleneck, generate more NAD⁺, and thus support rapid proliferation, even if it's less efficient in ATP production [120]. Consequently, cancer cells often exhibit a metabolic phenotype known as the Warburg effect, characterised by high rates of glycolysis and lactate production even in the presence of oxygen. Moreover, mitochondria-bound hexokinase, a key glycolytic enzyme, promotes apoptosis resistance, suggesting that glycolysis may protect tumour cells from apoptosis [121]. In line with this, Zheng et al. showed that Fusobacterium nucleatum infection elevates ANGPTL4 expression, which enhances glucose uptake and glycolysis in colorectal cancer cells, thereby supporting both bacterial colonisation and tumour survival [122]. Moreover, F. nucleatum outer membrane proteins Fap2 and RadD induce cell death in human T-lymphocytes [123].
In replenishing the intracellular NAD⁺ pool, NAMPT also plays a critical role. Accordingly, the NAMPT enzyme is overexpressed in many cancers, including glioma, lymphoma, breast, prostate, gastric cancer, and bladder cancer, typically linked to poor patient prognosis, advanced disease stage, aggressive tumour behaviour, and chemotherapy resistance [124, 125]. NAMPT can also induce stemness in glioma cells [126]. NAMPT is highly expressed in immunosuppressive tumour-associated macrophages (TAMs) and supports tumour immune evasion. Importantly, tumours with high NAMPT expression are more responsive to anti-PD-L1 therapy, as NAMPT drives PD-L1 expression via NAD⁺-dependent TET1 and STAT1/IRF1 signalling [127].
Blocking NAMPT with NAMPT inhibitors depletes NAD⁺, impairing vital processes, which leads to cancer cell death and attenuates tumour growth. NAMPT inhibition triggers apoptosis in acute myeloid leukaemia stem cells by disrupting lipid homeostasis while sparing normal hematopoietic stem cells [128]. Although early studies of NAMT inhibitors showed promise, clinical development has been limited by dose-related toxicities (especially thrombocytopenia) and modest effectiveness as standalone therapies [125]. Interestingly, the microbiome may be the cause of the low efficacy of NAMP inhibitors. Microbiome-mediated resistance to NAMPT inhibitors arises because gut bacteria possess a nicotinamidase (PncA) enzyme that converts NAM into NA. As a key substrate of the Preiss–Handler pathway via NAPRT, NA supports NAD⁺ synthesis. Consequently, this bacterial action bypasses the inhibited NAMPT enzyme in the host, especially if certain tumours amplify the NAPRT gene [129]. A diet high in nicotinamide precursors can strengthen this resistance, while antibiotics that reduce gut microbiota can potentially overcome it by eliminating the bacteria responsible for this NAD⁺ salvaging mechanism [130, 131]. Mycoplasma hyorhinis, a frequent contaminant of tumour tissues, has been shown to express PncA, thereby redirecting NAD synthesis from the NAMPT-dependent salvage pathway toward the Preiss–Handler pathway, bypassing pharmacological inhibition of NAMPT, and thus conferring resistance to NAMPT inhibitors. Beyond resistance to NAMPT inhibitors, the microbial rerouting of NAD precursors may enhance tumour cell adaptability to ploidy changes [132] and influence immune responses within the TME. Chronic M. hyorhinis infection activates NF-κB signalling, driving pro-inflammatory cytokine and chemokine expression, and enhancing proliferation, migration, and invasion of cancer cells [133, 134]. These findings underscore the need to consider microbial contributions, such as NAD rewiring, when designing and evaluating NAD-targeted therapies in cancer.
Tryptophan metabolites and cell death control
Dynamic interaction between host and microbiome makes tryptophan (Trp) metabolism a critical factor in shaping cell death in the TME. Trp is an essential dietary amino acid, serving not only in protein synthesis but also as a precursor for key metabolites. Trp is processed by both host and microbial enzymes. Cancer cells often deplete Trp through high activity of indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO), diverting it into the kynurenine pathway to support immune evasion. The hyperactivity of the kynurenine pathway elevates the kynurenine/tryptophan ratio, a marker of immune suppression and poor prognosis, as high kynurenine levels promote tumour immune evasion and lead to T-cell apoptosis [135]. Moreover, Trp deprivation halted cell growth and led to cell death in human pluripotent stem cells [136]. In mouse models of lung cancer, Trp supplementation or IDO inhibition enhanced CD8⁺ T-cell–mediated apoptosis of cancer cells, increased CD8⁺ T-cell infiltration into tumours, and slowed tumour growth. Trp potentiated anticancer activity of CD8^+^ T-cells by TRIP12 tryptophanylation and CD8^+^ T-cell surface PD-1 downregulation [137].
The shortage of Trp not only suppresses T-cell proliferation but also reshapes the microbiome. Many commensal gut bacteria are Trp auxotrophs that depend on environmental supply. For instance, the Ruminococcaceae species Faecalibacterium prausnitzii or Subdoligranulum variabile [138] or Actinobacteria, such as Collinsella and Olsenella [139], require external Trp to grow and to produce butyrate [140] that normally strengthens epithelial barrier function and regulates mucosal immunity [141, 142]. Patients who responded well to immunotherapy had higher levels of Ruminococcaceae and Collinsella in their gut, which correlated with stronger and longer-lasting treatment effects and increased T-cell infiltration in tumours [143, 144]. In a Trp-depleted TME, these beneficial bacteria may decline.
In the host, Trp can be used for protein synthesis (~ 1%) or can be metabolised through the kynurenine (~ 95%) (Fig. 2) or serotonin (~ 2%) pathways [145]. The microbiome adds another layer of complexity. The microbiota-dependent “pseudoendogenous” pathway converts Trp into a range of indole derivatives and other bioactive metabolites. Many gut bacteria, such as E. coli and Clostridium species, metabolise Trp into ligands for the aryl hydrocarbon receptor (AhR) and modulate mucosal immunity, gut barrier integrity, and inflammation [146]. Lactobacilli (such as Lactobacillus johnsonii) further shape this process by producing hydrogen peroxide, which suppresses host kynurenine metabolism through inhibition of IDO, the enzyme that catalyses kynurenine biosynthesis from Trp [147]. In cancer, kynurenine contributes to immune evasion and therapy resistance by signalling via AhR. Moreover, kynurenine can inhibit ferroptosis. Inside receiver cells, kynurenine activates an NRF2-dependent, AhR-independent pathway that upregulates expression of SLC7A11 (protein crucial for preventing ferroptosis, also known as xCT), thereby promoting further kynurenine import [148]. Kynurenine can also induce the expression of PD-1 in CD8^+^T-cells [149] and simultaneously can increase PD-L1 expression on tumour cells [150]. The binding of PD-L1 to PD-1 can lead to the apoptosis of activated T-cells, reducing the anti-tumour immune response [151]. Consequently, the anticancer effects of L. johnsonii can be mediated through the modulation of kynurenine in tumour cells. In papillary thyroid carcinoma (PTC), patients with lymph node metastasis show a significant reduction of L. johnsonii in tumour lesions. Gut colonisation with L. johnsonii increases its abundance in tumours, leading to inhibition of PTC growth and metastasis [152]. Conversely*, Streptococcus mutans* colonises OSCC tumours and drives excessive kynurenic acid (KYNA) production through its protein antigen c (PAc). KYNA promotes expansion of IL-1β–producing neutrophils, leading to CD8^+^T-cell exhaustion and apoptosis, immunosuppression, and OSCC progression. It also reduces the efficacy of PD-L1 and IL-1β blockade therapies, with high KYNA–AhR signalling correlating with poor patient survival [153].Fig. 2. Tryptophan metabolism and its connection with cell death. In host cells, tryptophan is metabolised through the kynurenine and serotonin pathways to generate bioactive metabolites or incorporated directly into protein synthesis. Cancer cells often deplete tryptophan through high activity of indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO), diverting it into the kynurenine pathway to support immune evasion. Lactobacilli produce hydrogen peroxide, which suppresses IDO and the host kynurenine production. A branch of the kynurenine pathway is involved in de novo NAD⁺ biosynthesis. Due to its role in the cell's antioxidant defence, NAD⁺ availability directly influences several forms of regulated cell death. In gut microbes, tryptophan is transformed into kynurenine, indoles, and derivatives such as tryptamine. These metabolites support intestinal barrier integrity and immune balance by activating the aryl hydrocarbon receptor (AhR), which promotes apoptosis of CD8^+^T-cells, generation of regulatory T-cell (Treg), and an immunotolerant microenvironment. In the tumour microenvironment, active AhR can promote immune evasion and drive cancerogenesis via repressing nucleotide excision repair (NER), apoptosis, and ferroptosis. Serotonin, 3-hydroxyanthranilic acid (3-HAA), indole‐3‐lactic acid (ILA), indole-3-propionic acid (IPA), trans-3-indoleacrylic acid (IDA), indole-3-pyruvate (I3P), and kynurenine were shown to inhibit ferroptosis. In contrast, indole-3-acetic acid (IAA) can promote ferroptosis
Dietary fibre serves as an important determinant of gut microbial Trp metabolism. In the gut, E. coli converts Trp into indole, while Clostridium sporogenes uses it in the Stickland pathway to regenerate NAD⁺ and produce indole‐3‐lactic acid (ILA) and indole-3-propionic acid (IPA). The fibre degrader Bacteroides thetaiotaomicron (indole producer, pectin degrader) breaks down pectin into monosaccharides that repress E. coli’s tnaA gene (tryptophanase), leaving more Trp available for Stickland fermenters [154]. ILA was shown to inhibit tumour cell proliferation, migration, and anti-apoptotic capabilities [155]. ILA also inhibits ferroptosis through activating the AhR/Nrf2 signalling pathway [156]. Lactobacillus johnsonii collaborates with Clostridium sporogenes to produce IPA. IPA modulates the stemness programme of CD8 + T-cells and improves responsiveness to immune checkpoint blockade at the pan-cancer level, including melanoma, breast cancer, and colorectal cancer [157]. IPA supplementation can inhibit ferroptosis and oxidative stress through the NRF2/GPX4 axis [158]. IPA also induces Ca^2+^-dependent apoptosis in *Candida albicans *[159]. Candida albicans acts as a driver of chronic inflammation, metabolic competition, and immune suppression in the TME, supporting tumour progression and potentially reducing therapeutic efficacy [160]. Several other indole derivatives produced by bacteria have been shown to act as potent ferroptosis inhibitors. 3-hydroxyindole, 6-hydroxyindole, 7-hydroxyindole, and indole-3-pyruvic acid suppress ferroptosis by scavenging free radicals and enhancing antioxidant gene expression [161, 162]. Trans-3-indoleacrylic acid (IDA), produced by Peptostreptococcus anaerobius, inhibits ferroptosis in tumour cells through a mechanism independent of lipid peroxidation. IDA treatment or implantation of P. anaerobius promotes colorectal cancer progression in both the xenograft model and ApcMin/+ mice via IDA–AhR–ALDH1A3 axis [163]. Additionally, indole and 7-hydroxyindole can reduce Pseudomonas aeruginosa virulence [164]. In contrast, Parabacteroides distasonis-derived indole-3-acetic acid (IAA) can promote ferroptosis by reducing FASN expression, decreasing the ratio of MUFAs to PUFAs, thereby increasing ferroptosis sensitivity in bladder cancer cells. P. distasonis is significantly decreased in bladder cancer patients, and its low abundance is strongly associated with poor prognosis [165]. Indoxyl sulphate has been shown to trigger apoptosis through mitochondrial ROS production [166, 167]. Indole-3-carboxaldehyde (ICA) is a gut microbe-derived tryptophan metabolite. ICA acts as a signalling molecule in the gut-brain axis, potentially improving stress and depression-like behaviours by influencing the AhR pathway. Supplementation with ICA or the ICA-producing strain Lactobacillus reuteri reduced neuroinflammation and promoted hippocampal neurogenesis via the AhR/SOCS2/NF-κB/NLRP3 pathway, alleviating pyroptosis [168]. Non-Trp-derived indole compounds that have a significant effect on cell death and inflammation include indole-3-carbinol (I3C), which originates primarily from the breakdown of glucosinolates in cruciferous vegetables, and its metabolite 3,3′-diindolylmethane (DIM). These metabolites induce apoptosis in cervical cancer cells by reducing BCL-2 expression [169] and trigger apoptosis in gastric adenocarcinoma cells via the mitochondrial pathway [170]. I3C can also reduce colitis by promoting the production of anti-inflammatory compounds like butyrate and modulating the gut microbiota [171].
Another active metabolites of Trp are serotonin and 3-hydroxyanthranilic acid (3-HAA). Serotonin and 3-HAA protect tumour cells from ferroptosis by acting as radical-trapping antioxidants that block lipid peroxidation. Monoamine oxidase degrades serotonin and abolishes its protective effect, while kynureninase promotes ferroptosis resistance via 3-HAA production. Conversely, 3-hydroxyanthranilate 3,4-dioxygenase consumes 3-HAA, restoring ferroptosis sensitivity, with its expression correlating with higher lipid peroxidation of cancer cells and better clinical outcomes of cancer patients [172]. On the other hand, 3-HAA suppresses NF-κB activation after T-cell receptor signalling by targeting PDK1, leading to apoptosis of activated Th2 cells [173] and monocyte-derived cells stimulated by interferon-gamma [174]. Th2 cells can suppress the growth of colon and pancreatic cancer by activating anti-tumour macrophage and eosinophil responses [175].
At least one bacterium species (such as Ruminococcus gnavus and Clostridium sporogenes) that encodes a tryptophan decarboxylase can be found in the gut microbiome of > 10% of humans. Such bacteria can produce tryptamine via tryptophan decarboxylation [176]. Tryptamine shows a dual role in cancer. In vitro, it inhibited proliferation, induced apoptosis, and reduced migration of prostate cancer cells (PC-3, LNCaP), with weaker effects on normal prostate cells (RWPE-1). In vivo, intratumorally injected tryptamine suppressed PC-3 xenograft growth, but systemic (i.p.) administration paradoxically promoted tumour progression [177]. This duality may be linked to its strong immunomodulatory activity: in an EAE mouse model, tryptamine reduced CD4⁺ and Th17 infiltration while expanding FoxP3⁺ regulatory T-cells (Tregs). Because Tregs suppress anti-tumour immunity and support tumour progression, tryptamine’s effects likely depend on the balance between its direct tumouricidal activity and its capacity to promote immune evasion [178]. Tryptamine was also shown to induce cell death with ultrastructural features of autophagy in neurons and glia [179]. The oral commensal Corynebacterium durum can convert tryptamine into an anticancer metabolite, durumamide A, an oxidative dimer of tryptamine. Durumamide A showed cytotoxicity across cancer cell lines (IC_50_: 25–35 μM) by targeting surviving (BIRC5) and triggering AIF-mediated apoptosis [180].
Short-chain fatty acids and bile acids
Short-chain fatty acids (SCFAs), including butyrate, acetate, and propionate, are produced by anaerobic bacteria through the fermentation of dietary fibres in the gut. Beyond serving as an energy source for colonocytes, SCFAs play a critical role in shaping the TME and regulating cancer cell fate by linking microbial metabolism of dietary fibre to both direct cancer cell death pathways and the modulation of anti-tumour immunity [181]. In the gut, Bacteroidetes commonly produce acetate, while propionate is generated by species such as Akkermansia muciniphila, Bacteroides vulgatus, and B. thetaiotaomicron. Butyrate is synthesised via acetyl-CoA, glutamate, lysine, or succinate pathways, mainly by clostridial clusters XIVa and IV, which include abundant species like *Faecalibacterium prausnitzii *[116]. Butyrate, in particular, functions as a histone deacetylase (HDAC) inhibitor, promoting apoptosis and cell-cycle arrest in cancer cells while sparing normal cells through the so-called “butyrate paradox.” The metabolic switch to aerobic glycolysis, known as the “Warburg effect,” limits cancer cells’ ability to oxidise butyrate. As a result, butyrate accumulates intracellularly, acting as an HDAC inhibitor and suppressing proliferation [182]. HDAC inhibitors butyrate and suberoylanilide hydroxamic acid (SAHA; vorinostat) trigger caspase-3 activation via the mitochondrial apoptotic pathway but can also induce caspase-independent autophagic cell death in multiple human cancer cell lines. This dual mechanism highlights their potential for treating cancers resistant to apoptosis [183]. Via HDAC inhibition, butyrate also potentiates lipoteichoic acid (LTA)-induced inflammasome activation [184]. Examples of bacteria that produce LTA include species of Staphylococcus, Streptococcus, and Bacillus, as well as many other Gram-positive microorganisms, such as *Enterococcus faecalis *[184]. LTA is a major component of their cell walls [185]. Butyrate and propionate also act as microbial danger signals, activating the NLRP3 inflammasome in human macrophages upon TLR stimulation [186]. Notably, butyrate-producing microbes, such as Butyricicoccus pullicaecorum, may counteract the oncogenic effects of p53 mutations in CRC by modulating chromosome segregation 1 (CSE1L) expression [187]. CSE1L is highly expressed in different cancers, correlating with advanced stage, poor prognosis, and worse patient outcomes. It also regulates chemotherapy-induced apoptosis, influencing cancer cell sensitivity to treatment [188, 189]. This suggests a potential microbiome-driven mechanism for restoring tumour-suppressive pathways and slowing cancer progression [190]. SCFAs can also influence other forms of regulated cell death: they modulate ferroptosis by affecting lipid metabolism. Sodium butyrate (NaB) enhances RSL3- and erastin-induced ferroptosis by increasing lipid peroxidation through downregulation of SLC7A11 and GPX4. This occurs via the FFAR2-AKT-NRF2 and FFAR2-mTORC1 axes in a cAMP-PKA-dependent manner. Functionally, NaB suppresses tumour growth, which can be reversed by mTORC1 activation or ferroptosis inhibition, highlighting its potential for colorectal cancer therapy through mTOR-dependent ferroptosis regulation [191]. Butyrate can also activate AMPK [192], which stabilises p53 protein by phosphorylating MDMX and inhibiting its activity [193]. Furthermore, AMPK senses cellular energy status and, when activated, phosphorylates and inhibits RIPK1 to suppress cell death and inflammation under nutrient stress. Nevertheless, prolonged stress overrides this inhibition, leading to cell death [194, 195]. GPR109A, the niacin/butyrate receptor, suppresses mammary tumorigenesis by inhibiting antiapoptotic signalling. Loss of GPR109A increases tumour incidence and lung metastasis in mice [196]. Accordingly, Lactiplantibacillus plantarum inhibited colon cancer cell proliferation as a function of its butyrogenic activity [197]. Butyrate and propionate were shown to induce apoptosis in lung and colorectal cancer cells [198, 199]. Moreover, SCFAs affect immune responses, with butyrate enhancing CD8⁺ T-cell function and regulatory T-cell differentiation, thereby indirectly shaping cancer cell survival or elimination [200].
Most circulating acetate in human plasma (50–200 μM) originates from gut microbial metabolism through the fermentation of dietary fibre. Indigestible polysaccharides are degraded by bacterial enzymes into hexoses (glucose, fructose), which are metabolised via glycolysis to pyruvate and then acetyl-CoA. This is converted to acetate through acetyl-phosphate and acetate kinase, generating ATP, or via the Wood–Ljungdahl pathway. There are metabolic links between different types of bacteria; for example, acetate produced by Bacteroidetes species can be utilised by Firmicutes to produce butyrate [116]. Interestingly, the Firmicutes/Bacteroidetes ratio was lower in patients with breast or colon cancer than in healthy controls [201, 202]. Non-consumed acetate is then released into the gut lumen. Tumour cells can absorb external acetate either through passive diffusion of acetic acid at low pH or active transport by monocarboxylate transporters (MCTs). Intracellular acetate is rapidly converted to acetyl-CoA by ACSS2 in the cytosol or ACSS1 in mitochondria. Cytosolic acetyl-CoA fuels biosynthesis of fatty acids, isoprenoids, sterols, ketones, and N-acetyl-glucosamines, which support cancer growth and the synthesis of cellular components, or serves as a substrate for lysine acetyltransferases to acetylate metabolites and proteins, including histones [118, 203]. For example, acetate induces expression of lipogenic genes ACACA and FASN by increasing H3K9, H3K27, and H3K56 acetylation, which enhances de novo lipid synthesis. This effect is reflected in hepatocellular carcinoma with high ACSS1/2 expression, where increased histone acetylation and lipid synthesis support cancer cell survival under hypoxia [204, 205]. Under glucose deprivation, AMPK phosphorylates ACSS2 at S659, driving nuclear translocation. In the nucleus, ACSS2 partners with TFEB, strongly promoting lysosomal biogenesis and autophagy, which are critical for tumour cell survival [206]. Acetate can also inhibit pyroptosis by attenuating inflammasome activation via GPR43-mediated Ca^2+^-dependent NLRP3 ubiquitination [207, 208]. On the other hand, acetate (as well as propionate, butyrate, formate, and pentanoate) can activate free fatty acid receptor 2 (FFAR2) and induce apoptosis through ERK signalling [209]. However, cancer cells can escape ERK-induced cell death by remodelling the ERK signalling. For example, pERK1/2 phosphorylates caspase-8 at S387, blocking its activation and thereby protecting cancer cells from caspase-8–mediated apoptosis [210].
The intestinal microbiome is also crucial for bile acid (BA) metabolism, converting primary to secondary BAs via microbial 7α-dehydroxylase and bile salt hydrolase (BSH) enzymes. BSH activity is widespread, especially in gram-positive bacteria such as Clostridium, Enterococcus, Bifidobacterium, Listeria, and Lactobacillus (e.g., L. plantarum). Deoxycholic acid (DCA), a secondary bile acid, influences host metabolism and may drive anti-apoptotic mechanisms by activating pathways such as EGFR (EGFR directly phosphorylates TNFR1 at Tyr360 and Tyr401, modulating NF-κB signalling and TNF-α–induced apoptosis and necroptosis) [211, 212], RelA [213], and Wnt5a/β-catenin [146, 214]. On the other hand, DCA can also trigger cell death by inducing oxidative stress, DNA damage, and mitochondrial dysfunction [215].
Krebs cycle metabolites lactate and ammonia
Excess NADH, which arises during hypoxia, mitochondrial dysfunction, or Warburg-like metabolic reprogramming, common in cancer, slows the electron transport chain (ETC) and inhibits succinate dehydrogenase (SDH, ETC complex II). This leads to the accumulation of both succinate and fumarate, which act as oncometabolites. Succinate stabilises hypoxia-inducible factor 1α (HIF-1α) by inhibiting prolyl hydroxylases and can also act extracellularly through the SUCNR1 receptor to promote inflammation and angiogenesis. Fumarate, in contrast, strongly modifies proteins through succination (e.g., of KEAP1), leading to NRF2 pathway activation and antioxidant responses. Succination also inhibits proteins such as the tumour suppressor PTEN and gasdermin D (GSDMD), with the latter suppressing pyroptotic cell death [216]. Both endogenous fumarate and exogenous dimethyl fumarate (DMF) succinate cysteine residues in GSDMD, preventing its interaction with caspases and subsequent activation [217]. In the absence of active GSDMD, caspase-1 can trigger apoptosis through the BID/caspase-9/caspase-3 pathway, occasionally followed by GSDME-mediated pyroptosis. Because GSDME is often silenced in tumours [73, 74], succinate may exert greater cytotoxicity in normal cells than in cancer cells. Indeed, succinate triggers pyroptosis in human umbilical vein endothelial cells (HUVECs) via inhibition of ATP5F1D [218], yet ATP5F1D inhibition does not induce pyroptosis in endometrial cancer cells [219]. Furthermore, succinate induces Ufsp2 transcription, promoting high-glucose–triggered pyroptosis in rat retinal Müller cells via NLRP3 inflammasome activation [220]. UFSP2 is frequently deleted in several human cancers (14 cancer types), and its absence promotes the growth of colon cancer cells and tumours [221]. Succinate also inhibits EGLN3, a key mediator of c-Jun–driven apoptosis [222]. EGLN3 suppresses gastric cancer malignancy by inhibiting JMJD8/NF-κB signalling [223]. Fumarate and succinate also exert epigenetic effects by inhibiting α-ketoglutarate–dependent dioxygenases, including histone and DNA demethylases. This inhibition causes a hypermethylated phenotype, altering the expression of genes controlling DNA repair, apoptosis, cell cycle, differentiation, and adhesion [216]. Together, succinate and fumarate integrate metabolic stress from NADH overload into pseudohypoxia, epigenetic remodelling, and survival signalling, driving tumour progression.
Gut microbes such as Bacteroides fragilis, Ruminococcus albus, Faecalibacterium prausnitzii, and Akkermansia muciniphila produce succinate through fermentation pathways. While some succinate is consumed by other gut bacteria, a portion is exported into the circulation, where it acts as a signalling metabolite. Circulating succinate can enter cancer cells through SLC13A3, a transporter directly regulated by β-catenin [224]. A multi-cohort metagenomic analysis of faecal DNA identified a gut bacterial signature—the ratio of succinate producers (Prevotellaceae + Veillonellaceae) to consumers (Odoribacteraceae + Clostridaceae)—as a key determinant of plasma succinate, which was higher in obese individuals. Changes in gut microbiota, whether via dietary weight loss or natural shifts, altered circulating succinate levels linked to microbial succinate metabolism [225]. Notably, Akkermansia muciniphila, a succinate producer from mucin degradation, is inversely associated with obesity-related metabolic disturbances and is enriched by prebiotics, metformin, and bariatric surgery [226]. This paradox can be partially explained by the trophic interaction between Akkermansia muciniphila and the butyrogenic gut commensal Anaerostipes caccae. Mucin degradation by A. muciniphila promotes A. caccae growth and butyrate production via the acetyl-CoA pathway. In turn, A. caccae alters A. muciniphila’s transcription, upregulating mucin degradation genes and downregulating ribosomal genes [227]. Clostridiales, such as A. caccae, are linked to lower colorectal tumour burden and are reduced in CRC patients. Oral administration of four Clostridiales strains (CC4) prevented and treated CRC in mice via CD8⁺ T-cell activation, with single strains such as Anaerostipes caccae or Roseburia intestinalis being even more effective. CC4 supplementation outperformed anti-PD-1 therapy, highlighting gut bacteria (particularly A. caccae) as a promising stand-alone cancer therapy [228]. Enterohemorrhagic E. coli (EHEC) is linked to CRC through its ability to induce DNA damage, inflammation, and chronic oxidative stress in the colon, which are known contributors to cancer development. EHEC can induce both apoptosis and autophagy in host cells, contributing to intestinal damage [229, 230]. Fumarate significantly increased EHEC virulence [231].
In cancer, high glycolytic flux converts most pyruvate into lactate rather than feeding it into the Krebs cycle (Warburg effect). This rerouting preserves mitochondrial intermediates for biosynthesis while regenerating NAD⁺ to sustain glycolysis under hypoxic or nutrient-stressed conditions. Consequently, tumours often exhibit high lactate levels. Lactate acts as both a metabolic fuel and a signalling molecule that fosters cancer cell survival and therapy resistance [232]. Some Lactate-producing bacteria, such as Lactobacillus iners and Staphylococcus, found within tumours can even enhance lactate production and contribute to cancer progression, rewiring tumour cell metabolism to fuel growth and resistance to cell death during treatments like chemotherapy and radiation [233, 234]. Lactate accumulation in tumours not only acidifies the microenvironment but also prevents glucose deprivation-induced parthanatos in gastric cancer cells [235] or melanoma cells [236] and suppresses apoptosis by stabilising HIF-1α via lysine lactylation at species-specific residues (K644 in mice and K12 in humans), enhancing protein stability by preventing VHL-mediated recognition [237]. HIF-1α promotes resistance to cell death via drug efflux pumps and accelerated DNA repair [238] and drives cancer resistance to cuproptosis [239]. Mitochondrial HIF-1α also protects against oxidative stress-induced apoptosis by reducing ROS levels and mitochondrial damage [240].
In healthy adults, lactate rarely accumulates in the colon, as it is consumed by other bacteria, including propionate- and butyrate-producers. Lactate, however, is a major fermentation product in breast-fed infants with Bifidobacterium-dominated microbiota. In adults, lactate accumulation is linked to dysbiosis, such as in severe colitis, often due to a deficiency of lactate-utilising bacteria [241]. These microbes, including species such as Anaerostipes caccae, Anaerobutyricum soehngenii (formerly Eubacterium hallii), and Veillonella parvula, convert lactate into metabolites like butyrate, propionate, and succinate [242]. In the context of the TME, Veillonella parvula can inhibit breast tumour growth and metastasis by selectively colonising tumours and reducing intratumoral lactate levels [243]. Proliferation-inhibiting, invasion-inhibiting, and apoptosis-promoting function of Veillonella parvula was also observed in oral squamous cell carcinoma [244]. Engineered E. hallii is being explored for use as a living biomaterial to convert intratumoral lactic acid into butyrate [245].
In addition to anaerobic glycolysis, cancer cells undergo metabolic reprogramming that heightens their reliance on glutamine. Glutamine supplies energy and carbon/nitrogen for the synthesis of amino acids, nucleotides, and fatty acids. The gut bacteria were shown to reshape amino acid metabolism. Compared to germ-free mice, conventional mice show higher portal vein glutamine but lower levels of many other amino acids (Arg, Asn, His, Ile, Leu, Met, Phe, Pro, Ser, Thr, Trp, Tyr, Val), indicating that microbes boost systemic glutamine while limiting the level of other amino acids [246]. Under hypoxia, glutamine fuels lipid synthesis via reductive carboxylation. This glutamine flux into α-ketoglutarate releases ammonia, leading to high ammonia levels (1–10 mM) in the TME, despite partial reuse by cancer cells [247]. Ammonia accumulation in the TME can become toxic to cancer cells when it exceeds buffering or recycling capacity. Excess ammonia raises lysosomal pH, disrupting lysosomal storage and causing ammonia reflux into mitochondria. This leads to mitochondrial damage and cell death, marked by lysosomal alkalization, mitochondrial swelling, and impaired autophagic flux [248]. Nevertheless, acidification of the TME by lactate can have a profound effect on ammonia toxicity. Mechanistically, increased glutaminolysis produces excess NH4⁺, which is normally exported from mitochondria and cleared from the microenvironment by proper perfusion. When perfusion is impaired, ammonia accumulates outside the cell. At physiological pH, ~ 2.2% of total ammonia exists as NH3, which can freely diffuse across the plasma membrane into the acidic cytosol. If NH3 influx surpasses NH4⁺ efflux, ammonia builds up inside the cell, alkalinizing acidic organelles, disrupting their function, and impairing cell growth and survival. At lower extracellular pH (6.8), only ~ 0.4% of ammonia exists as NH3, and the reversed pH gradient limits its entry into the cytosol. As a result, high extracellular ammonia has little effect under acidic conditions [247]. Consequently, high blood ammonia can be lethal to normal cells, such as CD8^+^ T-cells [249], but tumour cells are shielded by lactate-induced acidosis within the TME. CRC patients exhibit elevated serum ammonia and an ammonia-related gene signature, which is associated with T-cell apoptosis, weakened immune responses, poor prognosis, and resistance to immune checkpoint therapy [249]. The microbial involvement should be considered when assessing ammonia production and its systemic effects, as ammonia is the main nitrogen source in the gut, where many microbes both produce and utilise it, thereby influencing blood ammonia levels. Some microbes generate ammonia either by urease-mediated urea breakdown (e.g., Enterobacteriaceae, such as Klebsiellae, Veillonellaceae, Staphylococcus aureus, Mycobacterium tuberculosis, or H. pylori) or by proteolysis (e.g., Bacteroides, Clostridia, Fusobacterium) [250, 251]. On the other hand, some bacteria also consume ammonia, supporting detoxification. For example, a genetically engineered Lactobacillus plantarum strain efficiently converted ammonia into alanine, markedly lowering ammonia levels in both blood and feces [252]. Bell et al. have shown that promoting ammonia clearance attenuates apoptosis in T-cells and reactivates them, reduces tumour growth, and prolongs survival in CRC patients. Lowering ammonia also improves anti-PD-L1 efficacy, suggesting that targeting ammonia detoxification is a promising strategy to boost immunotherapy [249].
Polyamines, formate, and other bacterial metabolites
Polyamines, including putrescine, spermidine, and spermine, are small, positively charged metabolites produced by both host cells and the gut microbiota. Microbial polyamines boost colonic epithelial proliferation and shape macrophage differentiation. Colonisation of germ-free mice with wild-type, but not polyamine biosynthesis-deficient Escherichia coli, elevated intracellular polyamines in colonocytes, accelerating epithelial renewal. Additionally, bacterially derived putrescine promotes anti-inflammatory macrophages in the colon [253]. Polyamines are indispensable for cellular growth and survival, as they stabilise DNA and RNA, regulate ion channels, and support protein synthesis and cell cycle progression. When polyamine synthesis is reduced by mutations or pharmacological inhibition, cancer cells undergo senescence and apoptosis. Polyamine metabolism is tightly linked to the urea cycle through the intermediate ornithine, generated when arginase hydrolyses arginine. The primary polyamine produced from the urea cycle is putrescine, which is formed from ornithine by the enzyme ornithine decarboxylase (ODC). Putrescine is then further converted into other polyamines, such as spermidine and spermine. In cancer, arginase and ODC activity are often upregulated, diverting arginine toward polyamine production to sustain rapid proliferation [254]. During colorectal tumorigenesis, the urea cycle becomes progressively activated. This activation can be driven by the absence of gut microbes with ureolytic capacity (e.g. Bifidobacterium), leading to the accumulation of intestinal urea. Elevated urea not only promotes colorectal cancer initiation but also skews macrophages toward a pro-tumoral phenotype through increased polyamine production [255]. At the same time, microbial production of polyamines in the gut provides an additional source that contributes to elevated systemic levels. Together, these pathways create a metabolic network that strongly supports tumour growth and survival. In the CRC mouse model, reduced arginine catabolism in the intestinal microbiota leads to elevated arginine levels in the TME. This promotes nitric oxide (NO) production and polyamine synthesis in tumour tissues, driving angiogenesis, M2 macrophage polarisation, and activation of the Wnt/β-catenin pathway, ultimately accelerating CRC progression [256]. Fan et al. found that germ-free mice have elevated colonic polyamine levels. Mechanistically, Lactobacillus murinus secretes small RNAs in extracellular vesicles that suppress host polyamine metabolism by targeting key enzymes. Notably, CRC in mice and humans is associated with reduced bacterial small RNAs and increased polyamines. These findings reveal a microbiota-mediated mechanism controlling host polyamine metabolism [257].
Despite their growth-promoting functions, polyamines act as a double-edged sword. When their catabolism is activated, polyamines can become toxic to cancer cells. Catabolic enzymes such as spermidine/spermine N1-acetyltransferase (SSAT) and polyamine oxidases (PAOX/SMOX) convert polyamines into hydrogen peroxide (H₂O₂) and reactive aldehydes. These byproducts generate oxidative stress, cause DNA damage, and disrupt mitochondrial function, ultimately leading to apoptosis [258]. Cancer cells are often addicted to making polyamines, which they need to grow. Hyperactivated SSAT makes them waste polyamines, increasing their need for recycling through the enzyme methylthioadenosine phosphorylase (MTAP). Affronti et al. showed that MTAP inhibition combined with SSAT upregulation synergistically increases apoptosis in prostate carcinoma models and patient explants [259]. Moreover, Bi et al. identified polyamines as a ferroptosis promoter functioning in an H₂O₂-dependent manner. Iron overload activates Wnt/Myc signalling, upregulating ODC1 and boosting polyamine synthesis, creating a positive feedback loop that amplifies ferroptosis. Ferroptotic cells also release polyamine-rich extracellular vesicles, sensitising adjacent cells and spreading ferroptosis in tumours. Moreover, polyamine supplementation increases cancer cell sensitivity to radio- and chemotherapy by inducing ferroptosis. Since cancer cells often have elevated polyamine pools, targeting this pathway offers a promising therapeutic strategy [260].
During polyamine synthesis, formate can be generated as a byproduct. This pathway begins with the deamination of methionine to form SAM, which then donates a propylamine group to polyamine synthesis, creating 5-methylthioadenosine (MTA), which can be released or recycled into adenine, methionine, and formate. Formate is the simplest carboxylic acid anion, derived from formic acid, supporting purine and thymidine synthesis [261]. Elevated systemic formate (from microbiota or tumour metabolism itself) can therefore feed tumour growth. High formate levels have been linked to chemotherapy resistance in lung cancer and to certain bacteria, including Leuconostoc lactis and Eubacterium siraeum. In CRC, increased Fusobacterium nucleatum (including clinical isolates) similarly drives chemoresistance through formate. High production of formic acid, lactic acid, and acetic acid was also observed in a clinical isolate of *Bacteroides faecalis *[262]. Moreover, F. nucleatum produces hydrogen sulphide from sulphur-containing amino acids, promoting cancers like colorectal, liver, and lung cancer. In CRC cells, hydrogen sulphide increased inflammation, survival, and migration, while transcriptome analysis showed activation of autophagy and inflammatory pathways. In mice, F. nucleatum and L-cysteine accelerated tumour progression, upregulated autophagy genes, reduced probiotics, and enriched pathogenic microbes [263]. Some other gut bacteria produce hydrogen sulphide through cysteine desulfurase–catalysed degradation of cysteine. The first cysteine desulfurases were identified in Escherichia coli, and since then, similar reactions have been described in common colonic bacteria, including Clostridium, Enterobacter, Klebsiella, Streptococcus, and sulphate-reducing Desulfovibrio. Exogenous hydrogen sulphide was shown to inhibit apoptosis in colon cancer cells. Consistently, studies on gut microbiota in patients with colonic adenomas and CRC reveal increased abundance of sulphur-producing bacteria such as Bilophila, Desulfovibrio, Bilophila wadsworthia, and *F. nucleatum *[264].
Urolithin A (uroA) is a microbial metabolite produced from ellagic acid and ellagitannins by certain gut bacteria (e.g., Eggerthellaceae family, Enterocloster spp.). Inducible uroC dehydroxylase (ucd) operon is a key for uroA production. Metagenomic and ex vivo analyses confirm that only microbiota actively transcribing ucd generate uroA, explaining inter-individual differences in urolithin metabolism [265]. UroA has gained attention because it can directly influence cancer cell fate. For example, uroA triggers cell cycle arrest and apoptosis in colorectal cancer cells by suppressing BCL-2, upregulating expression of the pro-apoptotic proteins p53 and p21, and enhancing ROS production [266]. Moreover, uroA inhibited the interaction between p53 and MDM2, inhibiting MDM2-mediated p53 polyubiquitination and degradation [267]. Finally, by dampening pro-inflammatory signalling pathways, uroA reduces the tumour-supportive environment. Mechanistically, uroA promotes TFEB nuclear localisation, enhances mitophagy, and limits IL-6 and TNF-α production and inflammation from damaged mitochondria, ultimately inhibiting tumour growth in breast cancer models [268].
Microbes and cancer treatment
Modulation of cell death pathways is a key mechanism by which tumour cells develop drug resistance. In CRC, F. nucleatum activates autophagy by targeting TLR4, MYD88, and specific microRNAs, creating a molecular network that contributes to chemotherapy resistance [52, 269]. Analogously, Peptostreptococcus stomatis, enriched in CRC patients, activates the ERBB2-MAPK pathway, promoting tumour growth and reducing the efficacy of EGFR and BRAF inhibitors, thereby facilitating resistance to targeted therapies [270]. Other mechanisms include intratumoral Gammaproteobacteria (e.g., E. coli K12, Klebsiella pneumoniae, Klebsiella oxytoca, Mycoplasma hyorhinis, Salmonella enterica) metabolising gemcitabine into its inactive form. Accordingly, in a colon cancer mouse model, intratumoral Gammaproteobacteria induced resistance to gemcitabine [271]. Colbert et al. demonstrated that L-lactate produced by tumour-resident Lactobacillus iners reprograms the metabolism of cervical cancer cells, enhancing the Warburg effect and glycolysis, as well as glutamate and galactose metabolism, thereby driving resistance to chemoradiation. Similarly, lactic acid bacteria correlate with outcomes in colorectal, lung, head and neck, and skin cancers. These findings suggest that tumour-associated lactic acid bacteria drive lactate signalling–mediated resistance and represent potential therapeutic targets across cancers [272]. Moreover, Battaglia et al. observed tumour immune evasion mediated by cancer-associated fibroblasts (CAF) infiltration and immune exclusion in tumours with higher microbial diversity [273].
Based on these facts, it would seem rational to use antibiotics. Antibiotics can exert anticancer effects through both direct and indirect mechanisms. Directly, antibiotics, such as minocycline, tigecycline, and doxycycline, can impair mitochondrial protein synthesis due to the bacterial origin of mitochondria. This disruption of mitochondrial function reduces ATP production, induces oxidative stress, and triggers apoptosis in rapidly dividing cancer cells [274]. Indirectly, antibiotics can target tumour-associated bacteria that promote chemoresistance or tumour survival. For example, metronidazole reduced intratumoral F. nucleatum and suppressed cancer cell growth in CRC mouse models. Notably, human colorectal cancers colonised by F. nucleatum and its associated microbes, including Bacteroides, Selenomonas, and Prevotella, retain this microbial community in distal metastases, indicating stability of the tumour-promoting microbiome across primary and metastatic sites [275]. Notably, a key translational finding revealed that coadministration of the antibiotic ciprofloxacin effectively reversed gemcitabine resistance in mice [271].
On the other hand, several bacteria, including genetically modified strains of Clostridium, Salmonella, and Bacillus Calmette-Guerin (BCG), an attenuated strain of Mycobacterium bovis, are being investigated or used to trigger the elimination of cancer cells by directly causing cell death or by stimulating the immune system. BCG triggers caspase-independent cell death in carcinoma cells and promotes the release of an endogenous damage-associated molecular patterns (DAMPs) biomolecule HMGB1 [276]. HMGB1 triggers neutrophil extracellular trap (NETs) formation through interactions with Toll-like receptor 4 (TLR4) [277]. Hypoxic areas in solid tumours provide a niche for tumour-specific colonisation by anaerobic Clostridium species. Clostridium novyi-NT and C. sporogenes have shown promise in preclinical and early clinical models (NCT03435952, NCT0192468). Although their single-agent anti-tumour effects are modest, they can be used as a tumour-selective gene delivery system. Genetic engineering (via CRISPR–Cas9 or allele-coupled exchange) allows Clostridium to deliver therapeutic genes, such as prodrug-activating enzymes or express immunomodulators (e.g., IL-2, TNF-α), enhancing immunotherapy while reducing systemic toxicity [278]. Salmonella enterica can kill cancer cells through direct mechanisms, such as activating apoptosis and pyroptosis, and indirectly by stimulating the immune system and inhibiting tumour blood supply. Tumours contain both oxygen-poor and oxygen-rich regions, limiting the use of obligate anaerobes like Clostridium, which survive only in anoxic zones. In contrast, facultative anaerobes such as Salmonella enterica can colonise all tumour regions, including normoxic areas, making them more effective for targeting small and metastatic tumours. Its ability to selectively colonise tumours and deliver therapeutic agents also contributes to its effectiveness, making it a promising tool in bacterial-mediated cancer therapy [279, 280]. Moreover, Salmonella enterica serovar Typhimurium (S. typhi) selectively grows in tumours and reduces P-glycoprotein (P-gp) expression in cancer cells via its effector SipA through a pathway involving caspase-3. Mimicking this, gold nanoparticles coated with SipA were designed. These particles accumulate in tumours, lower P-gp expression, and enhance chemotherapy efficacy, overcoming multidrug resistance [281]. It was also demonstrated that Salmonella can deliver the cytosine deaminase gene to tumour tissue, where it remains functional, converting 5-fluorocytosine to the active drug 5-fluorouracil [282]. Salmonella can serve as a delivery vehicle for anticancer drugs or pro-apoptotic genes to target tumours [283]. Attenuated S. typhi shows potential as a cancer therapy by depleting asparagine in the TME via its asparaginase (ansB) activity, which suppresses tumour growth. However, this asparagine depletion also induces T-cell metabolic arrest by destabilizing c-Myc, limiting T-cell responses and the potential for immune memory or checkpoint blockade therapies. Using an S. typhi mutant with ansB deletion (∆ansB) restored T-cell function while still effectively reducing tumour growth [284]. The only FDA-approved bacterial therapy for cancer is BCG, which is used to treat non–muscle invasive bladder cancer (NMIBC). On April 22, 2024, the FDA approved nogapendekin alfa inbakicept-pmln (Anktiva, Altor BioScience, LLC) in combination with BCG for adults with BCG-unresponsive NMIBC with carcinoma in situ, with or without papillary tumours. The efficacy of Anktiva was evaluated in the QUILT-3.032 trial (NCT0302285).
Interestingly, some bacteria can directly take up extracellular DNA from their surroundings. This process represents one form of horizontal gene transfer (HGT); the exchange of genetic material between organisms outside of traditional parent-to-offspring inheritance. HGT is widespread among microbes but can also occur between microbes and higher organisms, including both microbe-to-eukaryote and eukaryote-to-microbe transfer. Cooper et al. advanced this concept by creating Acinetobacter baylyi strains capable of identifying oncogenic mutations in human DNA. This non-pathogenic bacterium can acquire DNA through HGT. The researchers modified it to gain drug resistance only when it acquired DNA containing a cancer-associated oncogene mutation, not the wild-type sequence. The engineered bacteria successfully detected their target in cell cultures and in mice with CRC, demonstrating promising diagnostic and tumour-targeting potential [285].
Conclusions and future direction: preserving the good, eliminating the bad
Several bacterial species that are highly abundant in tumours, such as Escherichia coli, Klebsiella pneumoniae, Staphylococcus aureus, Staphylococcus epidermidis, Pseudomonas species, Cutibacterium acnes [286], and even traditionally beneficial taxa such as Lactobacillus, illustrate the dual nature of host–microbe interactions. In their natural niches, these microbes often provide health-promoting functions. For example, E. coli contributes to vitamin K and vitamin B12 production [287] and iron metabolism in the gut, as recent research shows that enterobactin (a siderophore) produced by E. coli can benefit host iron levels in the intestine by carrying iron into host cells [288]. E. coli also maintains a friendly environment for its anaerobic neighbours by consuming oxygen that enters the gut [287]. S. epidermidis and C. acnes support skin barrier function, and Lactobacillus species help maintain gut homeostasis, enhance epithelial defence, and produce metabolites such as lactic acid that prevent pathogen overgrowth. However, when these same microbes colonise tumour tissues, their effects can shift dramatically toward tumour promotion. For example, the enterobactin-binding protein Lipocalin 2 and its receptor SCL22A17, overproduced by cancer cells, can redirect iron flow from E.coli to cancer cells, supporting their survival and growth in iron-scarce environments [289, 290]. Strikingly, Lactobacillus, generally regarded as a probiotic, can promote tumour cell survival by producing high levels of lactate that fuel glycolysis, rewire metabolism, suppress cell death of cancer cells, and confer resistance to chemoradiation [272]. Similarly, AhR is a ligand-activated transcription factor environmental sensor that, when activated, influences T-regulatory cells (Tregs) by promoting their homing and enhancing their immune-suppressive functions, a process linked to maintaining gut homeostasis [291]. In cancer, immune evasion and therapy resistance are supported by AhR signalling. AhR activity is significantly modulated by microbial products, including Trp metabolites, such as kynurenic acid [146]. In epidermal keratinocytes, AhR represses nucleotide excision repair and apoptosis and contributes to UV-induced skin carcinogenesis [292]. Both pharmacological inhibition and genetic deletion of AhR significantly enhance erastin-induced ferroptosis by downregulating SLC7A11 and increasing lipid peroxidation. Moreover, AhR ligand indole-3-pyruvate (I3P) protected cells from ferroptosis in an AhR-dependent manner. These findings suggest that AhR transcriptionally regulates SLC family genes to control ferroptosis, highlighting its key role in preventing lipid oxidation and maintaining cell survival under oxidative stress [293], which is undoubtedly desirable in the intestine but problematic in cancer treatment. Nevertheless, the impact of AhR activation in cancer is highly ligand-, cell type-, and context-dependent.
Furthermore, P-gp protects the intestinal epithelium by exporting xenobiotics and mediating immune–barrier communication. Recent findings show that specific gut microbes—mainly Clostridia and Bacilli—induce P-gp expression through synergistic actions of SCFAs and secondary BAs. In ulcerative colitis, reduced P-gp levels correlate with loss of these metabolites and impaired epithelial anti-inflammatory signalling. In cancer, high P-gp expression contributes to chemotherapy resistance. Physiological levels of butyrate significantly increased P-gp expression and activity in vitro, while acetate and propionate had no effect. Notably, butyrate alone was sufficient to induce P-gp expression in germ-free mice [294]. Therefore, therapeutic use of butyrate-producing bacteria in the TME should be combined with local administration of P-gp inhibitors to prevent enhanced drug efflux and maintain treatment efficacy. Together, these examples highlight that bacteria beneficial in the gut or skin may adopt tumour-supportive roles when present in the TME.
Tumour-resident microbes can drive cancer progression and drug resistance, yet systemic antibiotics are problematic because they disrupt the gut microbiome that supports immunity and metabolic health. Emerging strategies may focus on selective elimination of harmful bacteria in the TME while preserving gut commensals and their metabolites, supporting survival of intestinal cells during anticancer treatment, and preventing mucositis or acute radiation syndrome (e.g., indole-3-propionic acid (IPA) protects against radiation-induced toxicity by preserving acyl-CoA–binding protein function [295]). Wang et al. developed a liposome-encapsulated silver–tinidazole complex that selectively targets F. nucleatum. This approach triggered the release of tumour-specific microbial neoantigens and enhanced CD8⁺ T-cell infiltration, effectively transforming immune-cold tumours into immune-hot ones and promoting immune recognition and killing of cancer cells [296]. Other possible approaches include localised antibiotic delivery via nanoparticles and hydrogels [297, 298], bacteriophage therapy [299] tailored to tumour-associated strains (e.g., Fusobacterium nucleatum, Lactobacillus iners), inhibitors of bacterial enzymes such as cytidine deaminase that inactivate chemotherapeutics, or engineered bacteria supporting killing of cancer cells by the immune system. Immunotherapy is highly effective in blood cancers but less successful in solid tumours. To address this, Vincent et al. engineered probiotic E. coli that selectively colonise tumours and release synthetic antigens, effectively “tagging” them for chimeric antigen receptor (CAR) T-cells programmed to recognise these tags. This probiotic-guided CAR-T system safely directed immune attacks at solid tumours and enhanced therapeutic efficacy in breast and colon cancer models [300]. Additional options involve engineered butyrate- or IAA-producing bacteria improving butyrate and IAA bioavailability, providing localised anti-cancer effects [301] or improving the efficacy of anticancer therapy [302, 303]. Accordingly, Roseburia intestinalis sensitised colorectal cancer to radiotherapy through the butyrate/OR51E1/RALB axis, facilitating autophagy-related cell death [304].
A new promising therapeutic concept involves harnessing microbes and their metabolites to identify and modulate tumour sensitivity to specific forms of cell death. By systematically analysing microbial influences within the TME, it may become possible to selectively induce or amplify particular cell death pathways, thereby improving therapeutic precision. For instance, as mentioned in the previous text, F. nucleatum and S. typhi have been shown to enhance autophagy-related signalling, suggesting that their presence could be exploited to shift the balance and trigger autophagy-dependent cell death when combined with autophagy inducers under controlled conditions. Similarly, E. coli, Pseudomonas aeruginosa, polyamine metabolites, butyrate, or IAA can indicate tumour susceptibility to ferroptosis. On the other hand, the presence of bacteria expressing nicotinamidase (PncA) may confer resistance to NAMPT inhibitors and parthanatos. Moreover, microbes and metabolites such as E. coli, Chlamydia trachomatis, deoxycholic acid (DCA), succinate, and lactate are often associated with resistance to apoptosis or ferroptosis [305], and their elimination may support the anticancer effect of therapy. The main goal of anticancer therapy should be to trigger immunogenic cell death (ICD) in tumour cells to promote systemic antitumour immunity and the abscopal effect, while simultaneously minimising ICD in intestinal epithelial cells to prevent excessive inflammation and maintain the integrity of the gut barrier (Fig. 3). Unlike non-immunogenic death, ICD alerts and activates the immune system through the release of DAMPs, such as ATP, calreticulin, and HMGB1. These signals promote dendritic cell maturation, antigen presentation, and activation of cytotoxic CD8⁺ T-cells, thereby linking cell death to anti-tumour immunity. To optimise this approach, artificial intelligence (AI) and machine learning could be employed to integrate multi-omics data, including microbiome composition, metabolomic signatures, and tumour transcriptomics, to predict which microbial profiles sensitise or protect tumours from specific cell death pathways. Such models could guide the design of personalised microbe-guided cell death therapies. Ultimately, combining AI-driven prediction with microbial engineering holds the potential to reprogram tumour vulnerability with metabolic precision and spatial selectivity, offering a transformative strategy to overcome therapeutic resistance in cancer treatment.Fig. 3. Microbiome-based approaches to enhance immunogenic cell death of cancer cells and protect the gut barrier during cancer therapy. The main goal of anticancer therapy should be to trigger immunogenic cell death (ICD) in tumour cells to promote systemic antitumour immunity and the abscopal effect, while simultaneously minimising ICD in intestinal epithelial cells to prevent excessive inflammation, autoimmune attacks, and maintain the integrity of the gut barrier. The term ICD is used for all types of cell death that can initiate an adaptive immune response against dead-cell–associated antigens. A promising emerging strategy is to harness microbes and their metabolites to identify and modulate tumour sensitivity to specific cell death pathways. Systematic analysis of microbial influences within the tumour microenvironment (TME) could enable selective activation or enhancement of defined cell death mechanisms, improving the precision and efficacy of cancer therapy. CAF = Cancer Associated Fibroblasts; APC = Antigen-Presenting Cells
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Xie, W., Agarwal, S., & Yu, J. (2023). Ferroptosis: The vulnerability within a cancer monster. Journal of Clinical Investigation, 133, Article e 170027. 10.1172/JCI 17002710.1172/JCI 170027 PMC 1017883237183818 · doi ↗ · pubmed ↗
- 2Iftekhar, A., et al. (2021). Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nature Communications, 12, Article 1003. 10.1038/s 41467-021-21162-y 10.1038/s 41467-021-21162-y PMC 788103133579932 · doi ↗ · pubmed ↗
- 3Iske, J., et al. (2024). NAD⁺ prevents septic shock‑induced death by non‑canonical inflammasome blockade and IL‑10 cytokine production in macrophages. e Life, 12, Article RP 88686. 10.7554/e Life.8868610.7554/e Life.88686 PMC 1094259938372712 · doi ↗ · pubmed ↗
- 4Nelson, A. E., & Heske, C. M. (2025). Mechanisms of resistance to NAMPT inhibitors in cancer. Cancer Drug Resistance, 8, Article 18. 10.20517/cdr.2024.21610.20517/cdr.2024.216PMC 1205947640342733 · doi ↗ · pubmed ↗
- 5Zhou, L., et al. (2018). Faecalibacterium prausnitzii produces butyrate to maintain Th 17/Treg balance and to ameliorate colorectal colitis by inhibiting histone deacetylase 1. Inflammatory Bowel Diseases, 24, 1926–1940. 10.1093/ibd/izy 18210.1093/ibd/izy 18229796620 · doi ↗ · pubmed ↗
- 6Gao, J. et al. (2018). Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Frontiers in Cellular and Infection Microbiology, 8, Article 13. 10.3389/fcimb.2018.0001310.3389/fcimb.2018.00013 PMC 580820529468141 · doi ↗ · pubmed ↗
- 7Schneewind, O. & Missiakas, D. Lipoteichoic Acid Synthesis and Function in Gram-Positive Bacteria. in Biogenesis of Fatty Acids, Lipids and Membranes 1–18 (Springer, Cham, 2017). 10.1007/978-3-319-43676-0_17-2.
- 8Xu, Y. et al. (2020). Function of Akkermansia muciniphila in obesity: Interactions with lipid metabolism, immune response and gut systems. Frontiers in Microbiology, 11, Article 219. 10.3389/fmicb.2020.0021910.3389/fmicb.2020.00219 PMC 704654632153527 · doi ↗ · pubmed ↗