CDHR1 variants in a Japanese family with inherited retinal dystrophy and intrafamilial phenotypic variability
Toshiaki Hirakata, Dan Gao, Minami Oshima, Fumihiro Hara, Shintaro Nakao, Akira Murakami

TL;DR
A Japanese family with inherited retinal dystrophy shows varied symptoms despite shared CDHR1 gene variants, highlighting complex genetic effects.
Contribution
Identifies CDHR1 variants in a family with IRD and demonstrates significant intrafamilial phenotypic variability.
Findings
Three brothers with IRD showed different retinal degeneration patterns despite shared CDHR1 variants.
CDHR1 variants c.748C>A and c.2027T>A were identified, but their phenotypic implications remain complex.
The study emphasizes the need for cautious interpretation of genetic results in CDHR1-related retinal disease.
Abstract
To report a Japanese family with inherited retinal dystrophy (IRD) in which CDHR1 variants were identified, and to characterize the marked intrafamilial phenotypic variability. This retrospective case series included three brothers diagnosed with retinal dystrophy at Juntendo University Hospital. Comprehensive ophthalmic evaluations were performed, including best-corrected visual acuity (BCVA), Goldmann perimetry, fundus photography, fundus autofluorescence (FAF), optical coherence tomography (OCT), and full-field electroretinogram (ERG). Genetic testing was conducted using next-generation sequencing with an IRD gene panel. All three patients exhibited progressive visual decline with onset in their 40s–50s. Fundus examination revealed severe macular atrophy in two brothers (Cases 1 and 2), consistent with cone–rod dystrophy, whereas the youngest (Case 3) showed diffuse retinal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
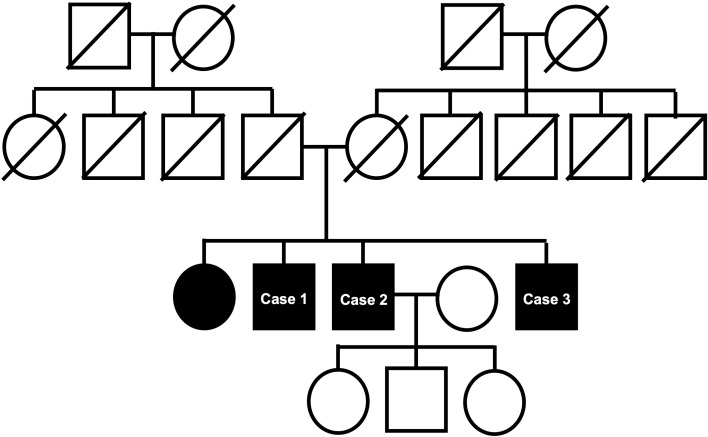 Figure 1
Figure 1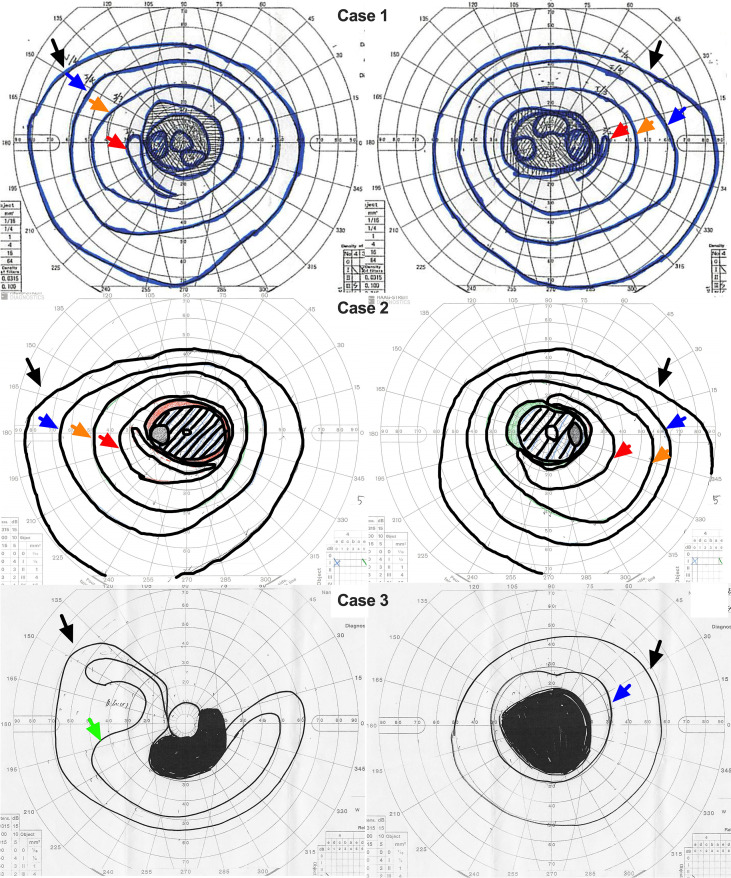 Figure 2
Figure 2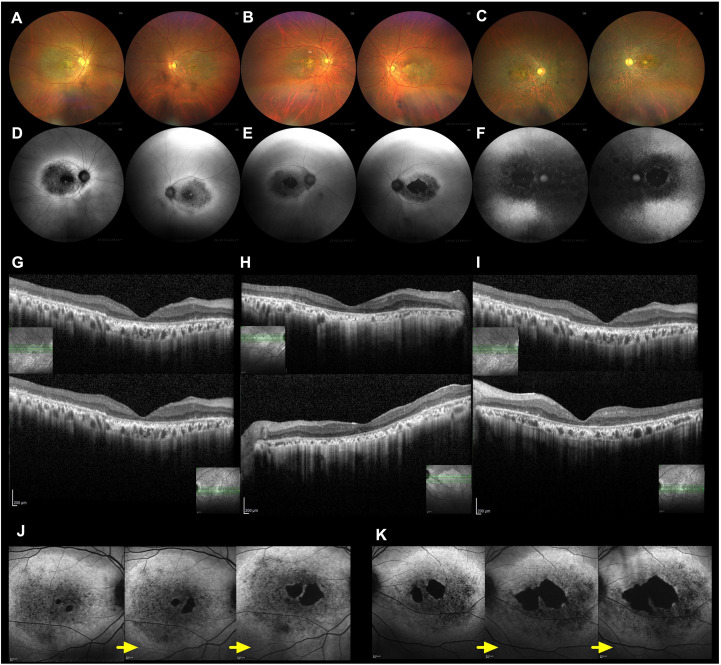 Figure 3
Figure 3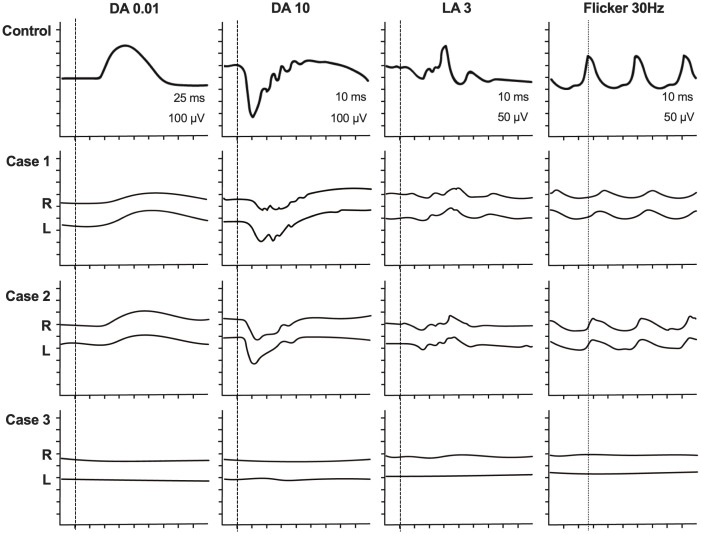 Figure 4
Figure 4| Gene | Nucleotide change (HGVS) | Zygosity | rsID | ClinVar dbSNP rsID | ACMG | ToMMo 61KJPN(# of homozygous individuals) | gnomAD v.4.1.0 | REVEL score | HGMD(Phenotype) | PMID | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 |
| c.748C>A p.(Pro250Thr) | heterozygous | − | − | VUS (PM2) | Not found | Not found | 0.415 | none | none |
| Case 2 |
| c.1738A>C p.(Asn580His) | heterozygous | rs199674847 | Conflicting classifications of pathogenicity Uncertain significance(5); Benign(1); Likely benign(1)(★) | VUS (BS1) | 0.007491(5) | Total:0.0001190 East Asian:0.004210 | 0.306 | CM157898(Cone dystrophy) | PMID:26161267 PMID:31213501 |
|
| c.748C>A p.(Pro250Thr) | heterozygous | − | − | VUS (PM2) | Not found | Not found | 0.415 | none | none | |
| c.2027T>A p.(Ile676Asn) | heterozygous | rs186486854 | Uncertain significance(★★) | VUS (BS1) | 0.002372(2) | Total:0.00008178 East Asian:0.002809(homozygotes:1) | 0.565 | PMID:24154662 PMID:30992995 PMID:33691693 PMID:33946315 PMID:33608557 | |||
| Case 3 |
| c.1738A>C p.(Asn580His) | heterozygous | rs199674847 | Conflicting classifications of pathogenicity Uncertain significance(5); Benign(1); Likely benign(1)(★) | VUS (BS1) | 0.007491(5) | Total:0.0001190 East Asian:0.004210 | 0.306 | CM157898(Cone dystrophy) | PMID:26161267 PMID:31213501 |
|
| c.392G>A p.(Arg131Gln) | heterozygous | rs752150870 | Uncertain significance(★★) | VUS (BS1) | 0.003173(1) | Total:0.00005771 East Asian:0.002052 | 0.375 | none | PMID:31213501 PMID:34073704 | |
|
| c.748C>A p.(Pro250Thr) | heterozygous | − | − | VUS (PM2) | Not found | Not found | 0.415 | none | none | |
| c.2027T>A p.(Ile676Asn) | heterozygous | rs186486854 | Uncertain significance(★★) | VUS (BS1) | 0.002372(2) | Total:0.00008178 East Asian:0.002809(homozygotes:1) | 0.565 | PMID:24154662 PMID:30992995 PMID:33691693 PMID:33946315 PMID:33608557 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRetinal Development and Disorders · Ocular Disorders and Treatments · Retinopathy of Prematurity Studies
Introduction
Autosomal recessive inherited retinal dystrophies (IRDs) are genetically heterogeneous and often associated with loss of function of cone and rod photoreceptors, potentially leading to blindness (1). Cadherin-related family member 1 (CDHR1) is a causative gene for retinal dystrophy, with an autosomal recessive form of inheritance (2, 3). The cadherin-related family member 1 (CDHR1), also known as protocadherin 21, or photoreceptor-specific cadherin, is a structural transmembrane photoreceptor protein that localizes at the base of cone and rod photoreceptor outer segments and plays an essential role in the maintenance of the structure and survival of photoreceptors (4). It is encoded by CDHR1, a gene located on chromosome 10q23.1 (5).
Cdhr1-deficient mice exhibit progressive outer retinal degeneration and early electroretinographic abnormalities involving both rod and cone pathways.
The Cdhr1-deficient mouse exhibits structural and functional evidence of progressive outer retinal degeneration at a slow rate (6). And, electroretinograms (ERGs) showed early functional deficits affecting both rod and cone photoreceptors. In humans, CDHR1 variants have been associated with a range of phenotypes, including cone–rod dystrophies (CRD) (7, 8), rod–cone dystrophies (RCD), retinitis pigmentosa (RP) (9), and late-onset macular degeneration (LOMD) (10, 11).
In this study, we report a Japanese family harboring a novel CDHR1 variant and exhibiting marked intrafamilial phenotypic variability, highlighting the complexity of genotype–phenotype correlations in CDHR1-associated retinal dystrophies and the importance of comprehensive clinical and genetic evaluation.
Methods
This was a retrospective case series of patients presenting to Juntendo University hospital with a diagnosis of IRDs. Three male siblings from the same Japanese family were included. Detailed medical history and comprehensive ophthalmic examinations were performed, including visual acuity, visual field test, fundus photography (CLARUS 500; Carl Zeiss Meditec Inc., Dublin, CA, USA), fundus autofluorescence (CLARUS 500; Carl Zeiss Meditec Inc., Dublin, CA, USA), optical coherence tomography (OCT, Spectralis; Heidelberg Engineering, Inc., Heidelberg, Germany), full-field electroretinography (full-field ERG, LE-4000, Tomey, Nagoya, Japan), were performed in accordance with ISCEV standards (12). Genetic analysis was conducted using next-generation system (NGS) with a gene panel. Pathogenicity of identified variants was interpreted with reference to established databases and ACMG guidelines. All CDHR1 variants were annotated based on the RefSeq transcript NM_033100.4. This study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Juntendo University (M08-0468). Written informed consent was obtained from all participants.
Results
Clinical characteristics
The family comprises four siblings, with the eldest being a sister followed by three younger brothers, of whom the three brothers were included in this study. Case 1 was a Japanese male in his late 60s who first noted night blindness in his early 50s. His past ocular history included laser in situ keratomileusis (LASIK) at the age 52 and bilateral cataract surgery at the age 60.
Case 2 was a Japanese male in his mid-60s who developed night blindness in his late 40s, followed by photophobia in his early 50s. His medical history included hypertension managed with medication, and prior surgeries for hemorrhoids and lumbar disc herniation. He was a former smoker, with a 15-cigarettes-per-day habit until age 57. His children were asymptomatic at the time of evaluation.
Case 3 was a Japanese male in his early 60s who experienced the onset of night blindness in his mid-40s, particularly under low-illumination or rainy conditions. He subsequently noted gradual progressive visual decline. He underwent bilateral cataract surgery in his late 50s, with the left eye procedure performed during an acute glaucoma episode.
The three brothers also have a sister who was diagnosed with IRD at another medical institution. Her symptoms began around age 50, and she currently has severe visual impairment and has been unable to renew her driver’s license since that time. The pedigree is shown in Figure 1.
Pedigree of the family with haplotype bars. Filled symbols represent affected individuals, and open symbols indicate unaffected individuals.
Vision and visual field
Goldmann perimetry (GP) of three cases was shown in Figure 2. At the initial visit, Case 1 (in his late 60s) had BCVA of 0.05 in the right eye and 0.06 in the left eye. GP revealed a central scotoma in both eyes.
Goldmann perimetry (GP) findings in all three cases. GP results for Case 1, Case 2, and Case 3 are shown from top to bottom. The right and left columns correspond to the right and left eyes, respectively. Cases 1 and 2 demonstrated central scotomas in both eyes, whereas Case 3 exhibited a central scotoma accompanied by peripheral visual field constriction. Black arrows indicate the visual field delineated by the V/4 isopter; a green arrow, the III/4 isopter; orange arrows, the I/3 isopter; blue arrows, the I/4 isopter; and red arrows, the I/2 isopter.
At the initial visit, Case 2 (in his late 50s) had decimal visual acuities of 1.2 in the right eye and 0.9 in the left eye. His vision gradually deteriorated over the following several years, reaching approximately 0.1 or less in both eyes by his early 60s. GP demonstrated a central scotoma in both eyes, similar to Case 1.
At the initial visit, Case 3 (aged 61 years) had visual acuities of 0.4 in the right eye and 0.05 in the left eye. GP revealed peripheral visual field constriction and generalized narrowing of the visual field.
Fundus images
Fundus photographs showed macular atrophy in both eyes of Cases 1 and 2, whereas in Case 3, retinal atrophy extended beyond the macula into the peripheral retina, accompanied by faint white spots and bone-spicule–like pigmentation in both eyes (Figures 3A–C). FAF demonstrated hypoautofluorescence in the macular area and surrounding hyperautofluorescence near the vascular arcades in both eyes of Cases 1 and 2 (Figures 3D, E). In contrast, FAF in Case 3 showed widespread hypoautofluorescence involving both the macula and peripheral retina (Figure 3F). OCT revealed loss of the outer retinal layers in the macula in all three cases (Figures 3G–I). Longitudinal FAF imaging clearly illustrated progressive macular degeneration in Case 2 (Figures 3J, K). Initially, visual acuity was preserved because the foveal retina remained intact; however, as foveal photoreceptor and retinal pigment epithelium (RPE) degeneration advanced, visual acuity gradually declined.
(A−C) shows the fundus photography of Cases 1, 2, and 3, respectively. (D−F) shows the fundus autofluorescence (FAF) images of Cases 1, 2, and 3, respectively. These images show macular degeneration and macular atrophy in both case 1 and 2, while not only macular atrophy and peripheral retinal degeneration in case 3. (G−I) Optical coherence tomography (OCT) images of Cases 1, 2, and 3, respectively (upper panels: right eyes; lower panels: left eyes). OCT images reveal outer retinal atrophy involving the retinal pigment epithelium (RPE) in the macular region of all three cases. (J, K) Longitudinal FAF images of Case 2 at ages 57, 58, and 61 years for the right and left eyes, respectively. In Case 2, the fovea was initially preserved; however, the hypoautofluorescent area corresponding to RPE atrophy gradually expanded over time.
Full-field electroretinogram
Full-field ERG demonstrated combined rod and cone dysfunction in both Cases 1 and 2. The dark-adapted dim flash ERG (0.01 cd·s·m^-^²; DA 0.01) showed markedly reduced responses, and the dark-adapted bright-flash ERG (DA 10) revealed severely attenuated a- and b-waves. The light-adapted cone ERG (LA 3) showed a markedly reduced amplitude, and the 30-Hz flicker ERG (LA 3, 30 Hz) exhibited both decreased amplitude and delayed implicit time. In contrast, Case 3 showed non-recordable responses under both dark- and light-adapted conditions, indicating advanced retinal dysfunction (Figure 4).
Full-field electroretinograms (ERGs) were recorded in all three cases. ERGs in the top row were shown as a normal control. In both case 1 and 2. Dark adapted dim flash ERG (0.01 cd•s•m-2; DA 0.01) showed a reduced response and bright flash ERG (DA 10) had severely reduced a-wave and b-wave. Light adapted cone ERG (LA 3) severely affected and 30Hz flicker ERG (LA 3, 30Hz) showed a reduction of amplitude and delayed implicate time. On the other hand, in case 3 full-field ERG showed un-detectable responses in both dark and light adapted conditions. Full-field electroretinograms (full-field ERGs) recorded in all three cases compared with a normal control (top row). In Cases 1 and 2, the dark-adapted dim-flash ERG (0.01 cd·s·m-²; DA 0.01) showed markedly reduced amplitudes, and the dark-adapted bright-flash ERG (DA 10) revealed severely attenuated a- and b-waves. The light-adapted single-flash ERG (LA 3) showed severely reduced cone responses, and the 30 Hz flicker ERG (LA 3, 30 Hz) demonstrated reduced amplitude and delayed implicit time. In contrast, Case 3 showed non-recordable responses under both dark- and light-adapted conditions.
Gene analysis
The summary of gene analysis is shown in Table 1. Following the evaluation of the sequencing data, we identified a heterozygous variant CDHR1 c.748C>A (p. Pro250Thr) in Case 1. Because only a single CDHR1 allele was affected, the molecular diagnosis in Case 1 remains inconclusive with respect to autosomal recessive CDHR1-associated IRD. In Cases 2 and 3, two heterozygous CDHR1 variants—c.748C>A (p.Pro250Thr) and c.2027T>A (p.Ile676Asn)—were detected. Because segregation analysis and phasing were not performed, the allelic configuration (cis or trans) could not be definitively determined, and therefore these variants are described as two heterozygous variants.
The CDHR1 c.748C>A (p.Pro250Thr) variant represents a novel variant that has not been previously reported. In addition, two variants of uncertain significance (VUS) were detected: a heterozygous PROM1 c.1738A>C (p.Asn580His) variant in Cases 2 and 3, and a heterozygous RP1 c.392G>A (p.Arg131Gln) variant in Case 3.
Discussion
We report three brothers with IRD in whom CDHR1 variants were identified, exhibiting marked phenotypic variability within the same family. Although the affected siblings showed overlapping clinical features, their genetic findings were not identical, and their clinical presentations differed substantially. Cases 1 and 2 showed similar findings characterized by severe macular atrophy, decreased visual acuity, and central scotoma, consistent with LOMD. However, the presence of night blindness and electroretinographic evidence of combined rod–cone dysfunction was atypical for isolated macular dystrophy. In contrast, Case 3 presented with fundus features resembling RP, including widespread retinal degeneration, bone-spicule pigmentation, subjective night blindness, and non-recordable full-field ERG responses, suggesting a rod–cone dystrophy phenotype.
Both CDHR1 variants identified in this study are currently classified as VUS according to ACMG/AMP guidelines. The c.2027T>A (p.Ile676Asn) variant was initially assigned PP3 based on in silico prediction; however, upon re-evaluation, the REVEL score (0.565) does not meet the threshold recommended by the ClinGen Sequence Variant Interpretation Working Group for supporting pathogenicity. Therefore, PP3 should not be applied. This variant shows relatively higher allele frequencies in East Asian populations, including the Japanese cohort, with reported homozygous individuals in public databases, supporting the application of BS1. Nevertheless, in Cases 2 and 3, c.2027T>A was detected together with a rare CDHR1 variant, c.748C>A (p.Pro250Thr), which is absent from population databases. Although parental segregation analysis was not available, the recurrent identification of these two heterozygous variants in affected siblings suggests a possible trans configuration. In this context, and consistent with the Japanese guideline of IRDs (13), PM3 supporting may be cautiously considered. Importantly, the available evidence remains insufficient to reclassify either variant as pathogenic or likely pathogenic, and both variants should be interpreted as VUS. Their contribution to disease pathogenesis remains hypothetical.
Case 1 carries only a single heterozygous CDHR1 c.748C>A (p.Pro250Thr) variant.
This monoallelic VUS is insufficient to establish CDHR1-associated autosomal recessive disease. Alternative explanations include an undetected second pathogenic allele (e.g., deep intronic variant, regulatory variant, or copy-number variant) or other genetic contributors not captured by our panel-based NGS. Therefore, Case 1 is considered genetically inconclusive despite its clinical resemblance to Cases 2 and 3.
In addition, beyond the REVEL score shown in Table 1, no additional in silico prediction tools were applied in this study. Given the absence of segregation analysis and comprehensive in silico evidence, the pathogenic relevance of the identified CDHR1 variants cannot be definitively established and should be interpreted with caution.
Variants in CDHR1 have been previously associated with a spectrum of autosomal recessive retinal dystrophies, including CRD (7, 8); RCD and RP (9, 14), and more recently, LOMD (10, 11). Intrafamilial phenotypic heterogeneity among individuals carrying identical CDHR1 variants has also been documented (7, 15). However, the mechanisms underlying these phenotypic differences remain poorly understood.
All three cases in this study shared several features: severe macular atrophy, late-onset symptoms, and self-reported night blindness, with ERG confirming dysfunction of both photoreceptor systems. These findings suggest that CDHR1-associated retinal dystrophy may present along a phenotypic continuum ranging from macular-predominant cone–rod dystrophy to generalized rod–cone dystrophy. Even in patients initially diagnosed with macular dystrophy, careful longitudinal follow-up is warranted to monitor for potential peripheral retinal involvement as the disease progresses.
An additional CDHR1 variant, c.2027T>A (p.Ile676Asn), was identified in Cases 2 and 3; however, this variant is currently classified as VUS, and its pathogenic role remains unclear. Other variants identified in this family, including those in PROM1 and RP1, may be relevant to retinal disease in general, but there is no direct evidence that they contribute to the observed phenotypic variability in the present cases.
Although non-Mendelian mechanisms such as genetic modifiers, oligogenic inheritance, or allelic expression imbalance have been proposed in IRD (16), these mechanisms remain speculative in the present family and are not supported by direct genetic or functional evidence. Accordingly, the phenotypic variability observed in this family should be interpreted with caution, and the possibility of additional undetected genetic or environmental factors cannot be excluded.
Although segregation analysis and functional validation were not performed, these findings expand the understanding of CDHR1 genotype–phenotype correlations. We propose that CDHR1 variants can manifest across a broad clinical spectrum, and that modifier variants or transcript-level regulation may underlie intrafamilial phenotypic diversity. Clinicians should consider CDHR1 variants in patients with macular dystrophy accompanied by night blindness or ERG evidence of rod–cone dysfunction.
This study has several limitations, including the absence of segregation analysis in the parents and the lack of in vitro or in vivo functional studies. Nevertheless, our findings contribute valuable insights into the expanding genotype–phenotype correlations in CDHR1-related retinal dystrophy.
Furthermore, even in cases with fundus findings suggestive of macular dystrophy, it is crucial to consider CDHR1 gene variants in clinical management when patients report night blindness or show evidence of rod-cone dysfunction on ERG. Taken together, our findings suggest that CDHR1-associated retinal dystrophy may manifest along a phenotypic continuum, ranging from macular-predominant disease to generalized retinal degeneration. However, the interpretation of genotype–phenotype correlations must be approached cautiously, particularly in the absence of segregation analysis and functional validation.
This study has several limitations, including the lack of segregation analysis, comprehensive structural variant detection, and in vitro or in vivo functional studies. Further investigations incorporating these approaches will be necessary to clarify the pathogenic relevance of the identified variants and to better understand the mechanisms underlying intrafamilial phenotypic variability in CDHR1-associated retinal dystrophy.
In conclusion, our findings illustrate marked intrafamilial phenotypic variability in a Japanese family with IRD in which CDHR1 variants were identified. These observations highlight the importance of integrating molecular, electrophysiological, and imaging analyses, as well as careful interpretation of genetic findings, for accurate diagnosis, prognostic assessment, and genetic counseling of patients with IRDs.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Heath Jeffery RC Mukhtar SA Mc Allister IL Morgan WH Mackey DA Chen FK . Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. (2021) 42:431–9. doi: 10.1080/13816810.2021.1913610, PMID: 33939573 PMC 8315212 · doi ↗ · pubmed ↗
- 2Nikopoulos K Avila-Fernandez A Corton M Lopez-Molina MI Perez-Carro R Bontadelli L . Identification of two novel mutations in CDHR 1 in consanguineous Spanish families with autosomal recessive retinal dystrophy. Sci Rep. (2015) 5:13902. doi: 10.1038/srep 13902, PMID: 26350383 PMC 4642573 · doi ↗ · pubmed ↗
- 3Duncan JL Roorda A Navani M Vishweswaraiah S Syed R Soudry S . Identification of a novel mutation in the CDHR 1 gene in a family with recessive retinal degeneration. Arch Ophthalmol. (2012) 130:1301–8. doi: 10.1001/archophthalmol.2012.1906, PMID: 23044944 PMC 3799916 · doi ↗ · pubmed ↗
- 4Rattner A Smallwood PM Williams J Cooke C Savchenko A Lyubarsky A . A photoreceptor-specific cadherin is essential for the structural integrity of the outer segment and for photoreceptor survival. Neuron. (2001) 32:775–86. doi: 10.1016/S 0896-6273(01)00531-1, PMID: 11738025 · doi ↗ · pubmed ↗
- 5Yagi T Takeichi M . Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes Dev. (2000) 14:1169–80. doi: 10.1101/gad.14.10.1169, PMID: 10817752 · doi ↗ · pubmed ↗
- 6Yusuf IH Mc Clements ME Mac Laren RE Charbel Issa P . Deep phenotyping of the Cdhr 1(-/-) mouse validates its use in pre-clinical studies for human CDHR 1-associated retinal degeneration. Exp Eye Res. (2021) 208:108603. doi: 10.1016/j.exer.2021.108603, PMID: 33964272 · doi ↗ · pubmed ↗
- 7Haque MN Kurata K Hosono K Ohtsubo M Ohishi K Sato M . A Japanese family with cone-rod dystrophy of delayed onset caused by a compound heterozygous combination of novel CDHR 1 frameshift and known missense variants. Hum Genome Var. (2019) 6:18. doi: 10.1038/s 41439-019-0048-8, PMID: 30992995 PMC 6459921 · doi ↗ · pubmed ↗
- 8Cohen B Chervinsky E Jabaly-Habib H Shalev SA Briscoe D Ben-Yosef T . A novel splice site mutation of CDHR 1 in a consanguineous Israeli Christian Arab family segregating autosomal recessive cone-rod dystrophy. Mol Vis. (2012) 18:2915–21., PMID: 23233793 PMC 3519373 · pubmed ↗