LAV-BPIFB4 reverses progeria-associated cardiac aging by restoring diastolic function and reducing senescence
Unbin Chae, Young-Ho Park, Taeho Kwon, Kyung-Sook Chung, Sun-Uk Kim

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
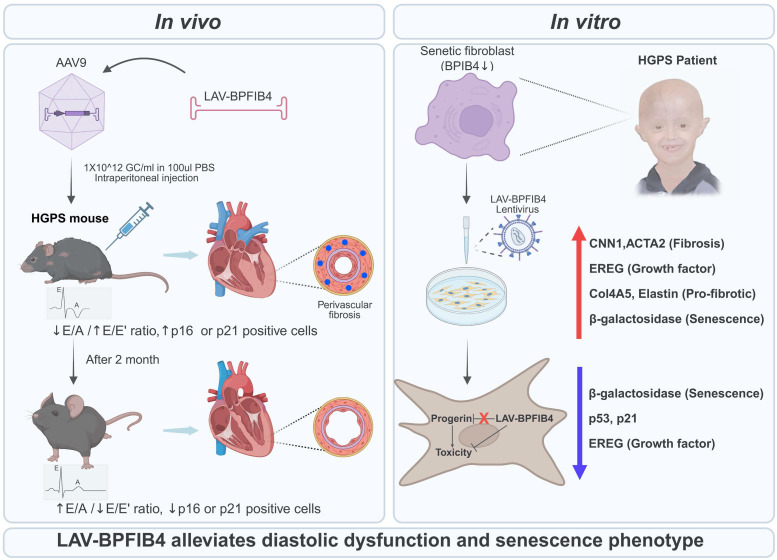 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNuclear Structure and Function · Renal and related cancers · Telomeres, Telomerase, and Senescence
A recent study published in Signal Transduction and Targeted Therapy by Yan Qiu demonstrated that activation of a longevity gene can partially reverse cardiac damage associated with Hutchinson-Gilford progeria syndrome (HGPS) 1. HGPS is primarily caused by a recurrent point mutation (c.1824 C>T, often designated as G608G) in exon 11 of the LMNA gene, which occurs de novo in most patients 2, 3. This mutation causes abnormal alternative splicing, impairing the function of a nuclear membrane protein called mutant lamin A or progerin. Progerin accumulates in the cell nucleus, disrupting nucleocytoskeletal coupling and contributing to characteristic symptoms of HGPS, including clinical features resembling premature aging, skeletal deformities, and shortened lifespan. Among the various systemic manifestations, cardiovascular dysfunction, particularly progressive atherosclerosis and cardiac fibrosis, represents the leading cause of mortality in HGPS patients 4. Consequently, the cardiovascular system has been identified as a primary therapeutic target in progeria research. Over the past decade, extensive efforts have been devoted to improving cardiac outcomes in HGPS, ranging from conventional pharmacological strategies aimed at mitigating vascular pathology 5 to the recent development of CRISPR-based approaches designed to correct the underlying LMNA mutation and selectively eliminate progerin expression 6, 7. These advances underscore the growing focus on restoring cardiovascular integrity as a central objective of effective therapeutic intervention in HGPS.
Recent studies have introduced a promising paradigm for drug target discovery grounded in the genetics of human extreme longevity 8. By systematically analyzing the genomes of individuals with exceptional lifespans, researchers have uncovered rare variants within evolutionarily conserved, aging-related pathways. These variants have undergone rigorous functional validation in both cellular and animal models, ultimately establishing a framework for identifying genetic alterations as molecular targets for the development of therapeutics aimed at prolonging healthspan. A compelling example of this approach is the BPI Fold Containing Family B Member 4 (BPIFB4) gene, particularly the longevity-associated variant (LAV; Ile229Val/Asn281Thr/Leu488Phe/Ile494Thr), which has been identified in the homozygous state in supercentenarians, individuals living beyond 110 years of age in good health 9. Enhanced expression of LAV-BPIFB4 in senescent vascular cells and aged mice not only restores cardiac function and vascularization but also confers substantial anti-aging benefits, thereby mechanistically linking longevity genetics to translational therapeutic applications 10. Notably, HGPS cells exhibited markedly reduced endogenous BPIFB4 expression, likely attributable to progerin-mediated suppression. Building on these insights, the present study posits that restoring BPIFB4 function may effectively counteract progerin-induced toxicity and ameliorate the cardiovascular dysfunction characteristic of HGPS.
The authors conducted experiments using 26-week-old HGPS mice that exhibited growth retardation compared to age-matched control mice (Figure 1) 1. Three-dimensional echocardiographic examinations revealed that untreated HGPS mice spontaneously developed left ventricular diastolic dysfunction between 7 and 8 months of age, characterized by a decreased E/A ratio and an elevated E/E' ratio. Conversely, HGPS mice receiving a single intraperitoneal injection of AAV9-LAV-BPIFB4 showed significant recovery of left ventricular diastolic function within 2 months post-transduction, with 2.5-fold enhancement of cardiac BPIFB4 protein expression. Histological analyses demonstrated that AAV9-LAV-BPIFB4 specifically reduced perivascular fibrosis, but not interstitial fibrosis, increased the number of coronary arterioles, and enhanced vascular smooth muscle cell coverage. Two months post-transduction, aortic tissue harvested from 8-month-old progeria mice revealed no detectable atherosclerotic lesions, consistent with the early-stage nature of this HGPS model rather than a direct anti-atherosclerotic effect of LAV-BPIFB4 1. Transduction of LAV-BPIFB4 into HGPS patient-derived fibroblasts led to reduced senescence and fibrotic markers, including calponin-1 (CNN1), smooth muscle α-actin (ACTA2), epiregulin (EREG), Col4A5, elastin, and β-galactosidase, without affecting progerin protein accumulation 1. This was achieved through suppression of the p53-p21 axis and modulation of EGFR-mediated fibrotic signaling.
The novelty of this study lies in identifying LAV-BPIFB4 as a therapeutic modality mechanistically distinct from progerin-elimination approaches 1. Rather than directly targeting progerin for degradation, LAV-BPIFB4 functions as a cellular resilience activator, restoring nucleolar integrity and function encompassing ribosome biogenesis, ribonucleoprotein assembly, and ribosomal protein interactions 1. Through this nucleolar-centric mechanism, LAV-BPIFB4 attenuates progerin-induced nucleolar and nuclear stress and nuclear dysfunction, thereby preserving diastolic performance, reducing cardiac fibrosis, and mitigating age-related senescence.
Importantly, this mechanism complements progerin-suppression strategies by enhancing cellular resilience rather than directly reducing progerin levels. While gene-editing interventions correct LMNA mutations or lower progerin burden, LAV-BPIFB4 reinforces nucleolar integrity, ribosome biogenesis, and stress-response capacity 1. Together, these complementary mechanisms may provide a multi-layered cardioprotective strategy for stabilizing the vulnerable myocardial environments.
In summary, this groundbreaking study reveals that LAV-BPIFB4, a protective variant identified in supercentenarian genetics, reverses cardiac dysfunction in progeria through a resilience-enhancing mechanism rather than direct progerin elimination. By selectively mitigating perivascular fibrosis and fortifying cardiomyocyte stress tolerance while preserving progerin expression, LAV-BPIFB4 establishes a fundamentally new therapeutic paradigm. These findings underscore that the systematic interrogation of human exceptional longevity genetics reveals multiple protective alleles with significant translational potential, not only for rare genetic aging diseases but also for age-related cardiovascular pathologies in the general population.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Qiu Y Cattaneo M Maciag A Puca AA Madeddu PA longevity-associated variant of the human BPIFB 4 gene prevents diastolic dysfunction in progeria mice Signal Transduct Target Ther 2025103144102270210.1038/s 41392-025-02416-3PMC 12480688 · doi ↗ · pubmed ↗
- 2Eriksson M Brown WT Gordon LB Glynn MW Singer J Scott L Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome Nature 200342329381271497210.1038/nature 01629 PMC 10540076 · doi ↗ · pubmed ↗
- 3De Sandre-Giovannoli A Bernard R Cau P Navarro C Amiel J Boccaccio I Lamin a truncation in Hutchinson-Gilford progeria Science 200330020551270280910.1126/science.1084125 · doi ↗ · pubmed ↗
- 4Chang W Wang Y Luxton GWG Ostlund C Worman HJ Gundersen GG Imbalanced nucleocytoskeletal connections create common polarity defects in progeria and physiological aging Proc Natl Acad Sci U S A 20191163578833080875010.1073/pnas.1809683116 PMC 6397528 · doi ↗ · pubmed ↗
- 5Gordon LB Shappell H Massaro JD'Agostino RB Sr Brazier J Campbell SE Association of Lonafarnib Treatment vs No Treatment with Mortality Rate in Patients with Hutchinson-Gilford Progeria Syndrome JAMA 20183191687952971016610.1001/jama.2018.3264 PMC 5933395 · doi ↗ · pubmed ↗
- 6Chae U Yang HJ Kim H Lee SH Lee DG Koo JY Precise progerin targeting using Rfx Cas 13d: A therapeutic avenue for Hutchinson-Gilford progeria syndrome Mol Ther 20253343944134051866710.1016/j.ymthe.2025.06.017PMC 12432902 · doi ↗ · pubmed ↗
- 7Olsen FJ Gordon LB Smoot L Kleinman ME Gerhard-Herman M Hegde SM Progression of Cardiac Abnormalities in Hutchinson-Gilford Progeria Syndrome: A Prospective Longitudinal Study Circulation 2023147178243727625410.1161/CIRCULATIONAHA.123.064370 · doi ↗ · pubmed ↗
- 8Zhang ZD Milman S Lin JR Wierbowski S Yu H Barzilai N Genetics of extreme human longevity to guide drug discovery for healthy ageing Nat Metab 20202663723271953710.1038/s 42255-020-0247-0PMC 7912776 · doi ↗ · pubmed ↗