Phenanthrene-like Benzodichalcogenophenes: Synthesis, Electrochemical Behavior and Applications
Valentina Pelliccioli, Serena Arnaboldi, Silvia Cauteruccio

TL;DR
This paper reviews the synthesis, electrochemical behavior, and applications of phenanthrene-like benzodichalcogenophenes in materials chemistry.
Contribution
The paper provides a comprehensive review of isomeric benzodichalcogenophene systems and their tailored properties for functional materials.
Findings
Isomeric benzodichalcogenophenes exhibit diverse structural and electronic properties based on ring orientation.
Tailored functionalization strategies allow for fine-tuning of their intrinsic properties.
These systems have applications in optoelectronics, supramolecular chemistry, and biological studies.
Abstract
Benzodichalcogenophenes represent a valuable class of organic π-conjugated systems that have been investigated in a plethora of cutting-edge applications in the field of materials chemistry. Isomeric benzodifuran (BDF), benzodithiophene (BDT) and benzodiselenophene (BDS) analogs of phenanthrene, in which the two heteroaromatic rings are ortho-fused onto a benzene ring, represent convenient frameworks as functional materials in organic electronics. The orientation of the two condensed heteroaromatic rings with respect to the central benzene ring provides diverse structural isomers, which significantly differ in degrees of curvature, electronic and electrochemical properties. Furthermore, tailored modification and functionalization strategies enable fine-tuning of their intrinsic properties, leading to unique systems. This review offers a comprehensive overview of synthetic methodologies…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
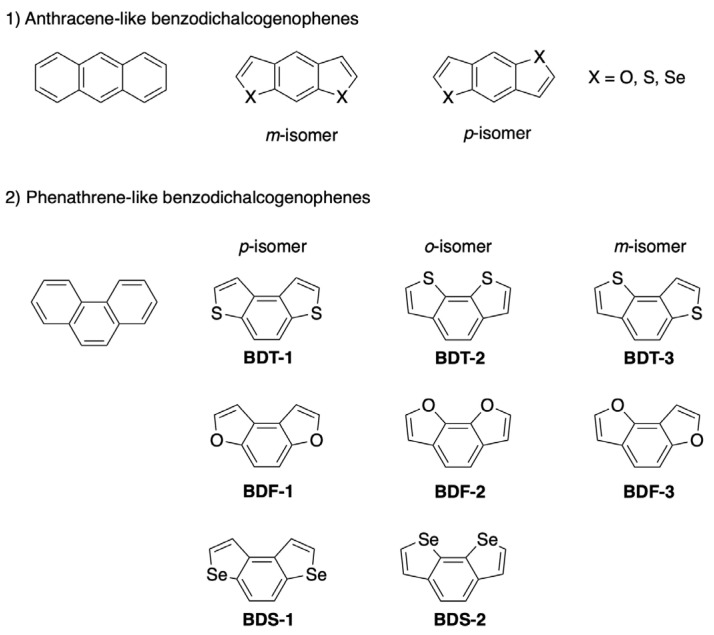 Figure 1
Figure 1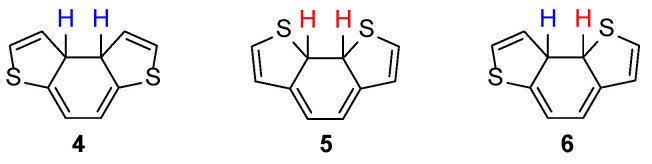 Figure 2
Figure 2 Figure 3
Figure 3 Figure 4
Figure 4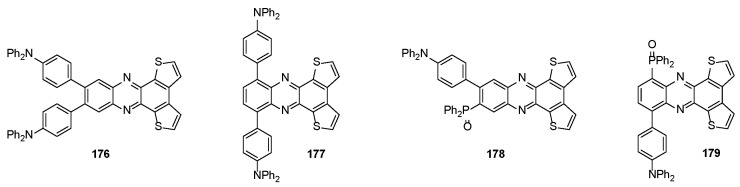 Figure 5
Figure 5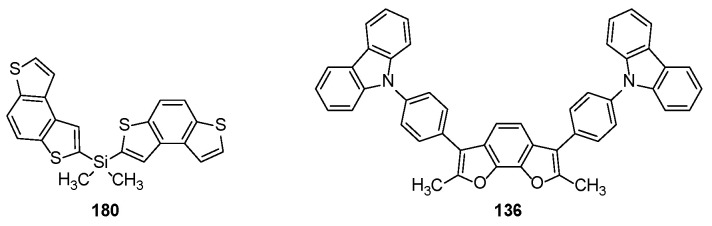 Figure 6
Figure 6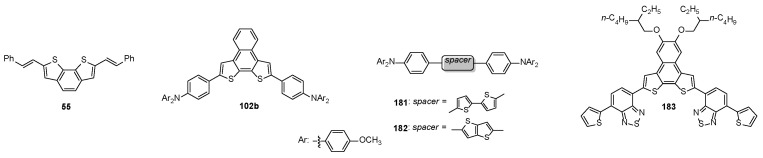 Figure 7
Figure 7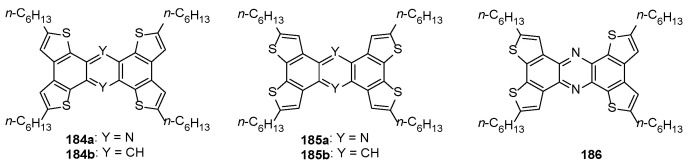 Figure 8
Figure 8 Figure 9
Figure 9 Figure 10
Figure 10 Figure 11
Figure 11 Figure 12
Figure 12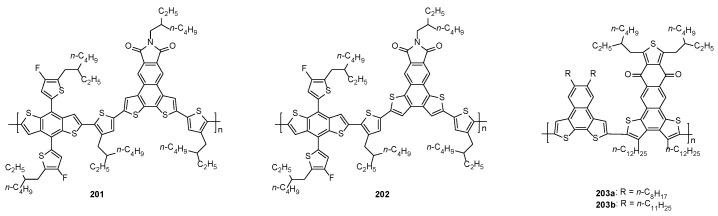 Figure 13
Figure 13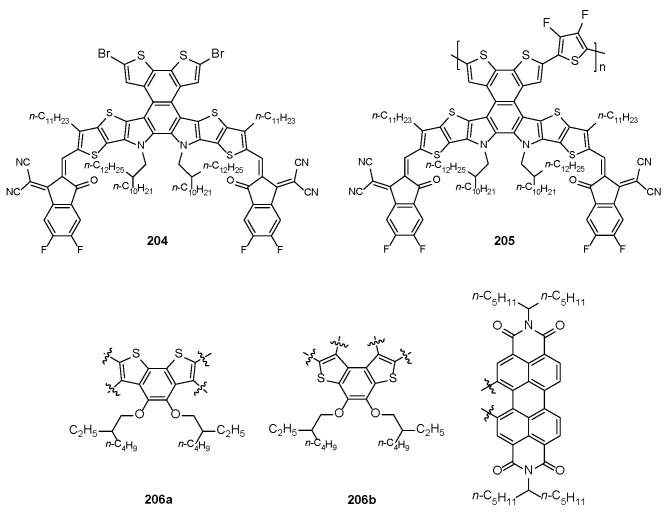 Figure 14
Figure 14 Figure 15
Figure 15 Figure 16
Figure 16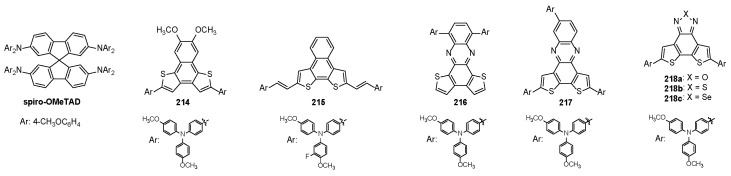 Figure 17
Figure 17 Figure 18
Figure 18 Figure 19
Figure 19 Figure 20
Figure 20 Figure 21
Figure 21 Figure 22
Figure 22 Figure 23
Figure 23 Figure 24
Figure 24 Figure 25
Figure 25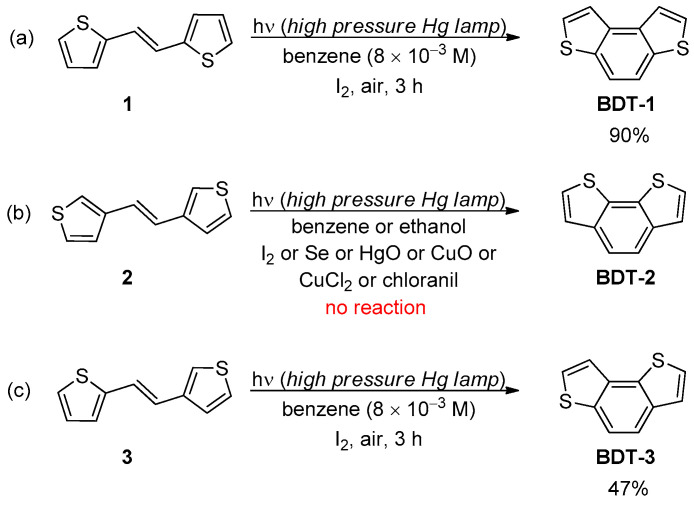 Figure 26
Figure 26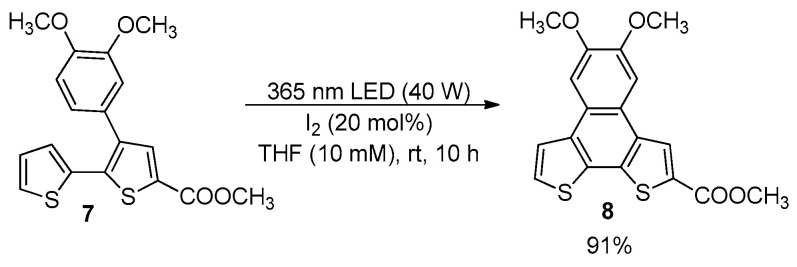 Figure 27
Figure 27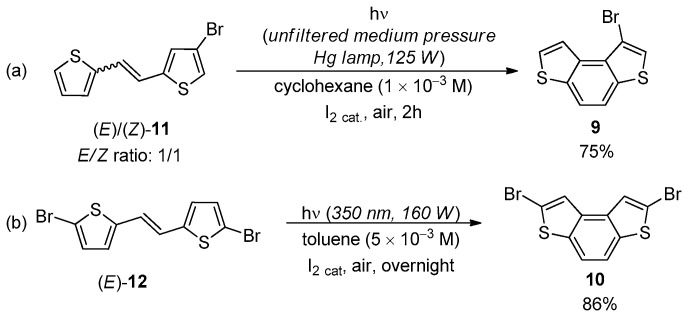 Figure 28
Figure 28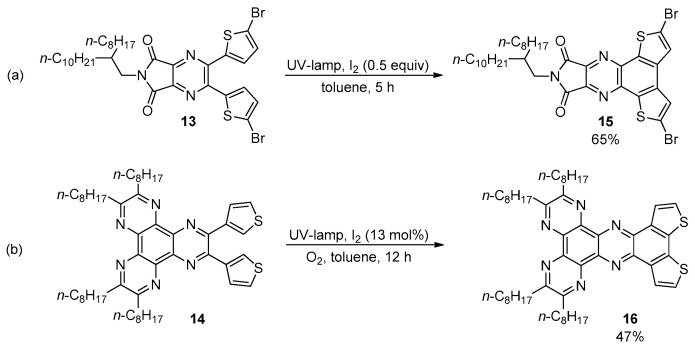 Figure 29
Figure 29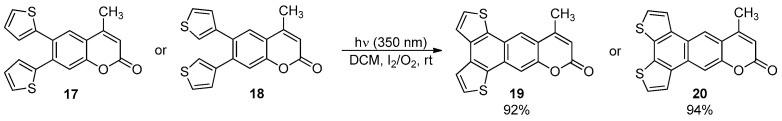 Figure 30
Figure 30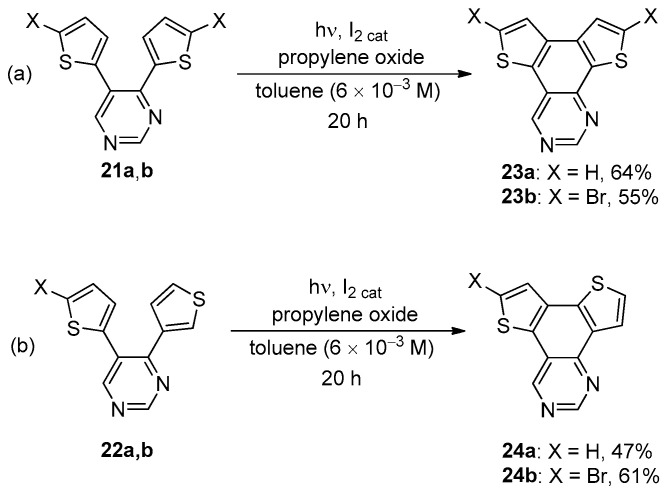 Figure 31
Figure 31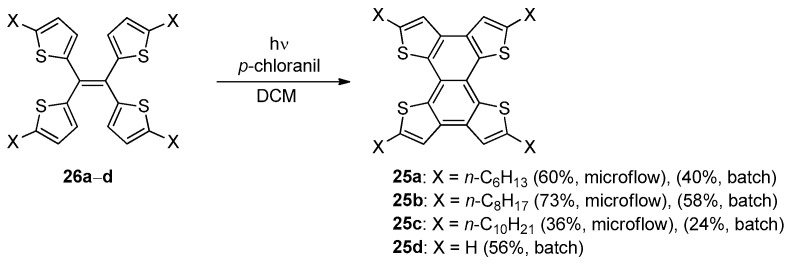 Figure 32
Figure 32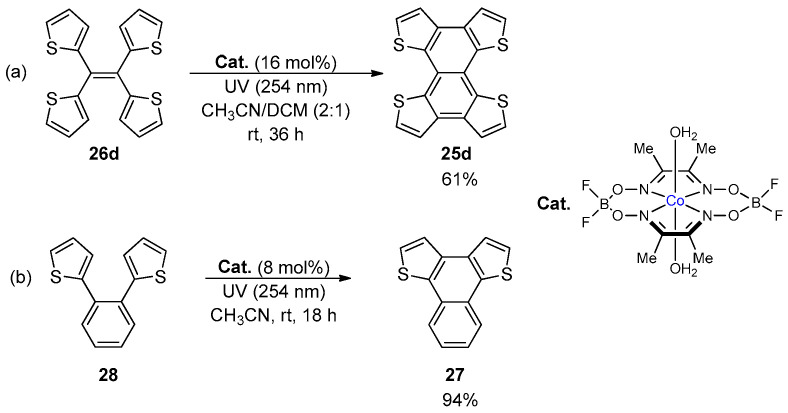 Figure 33
Figure 33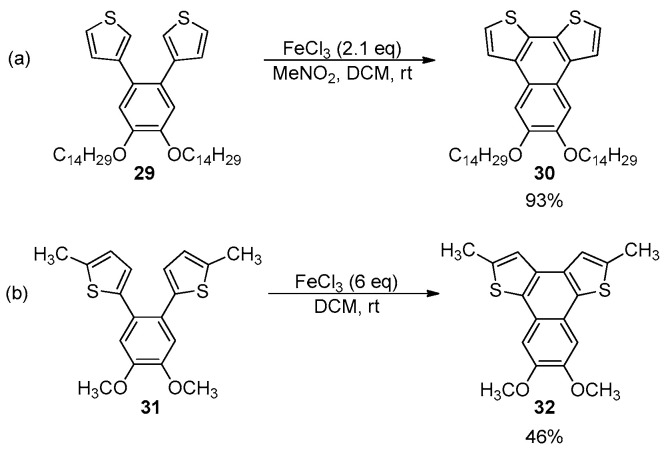 Figure 34
Figure 34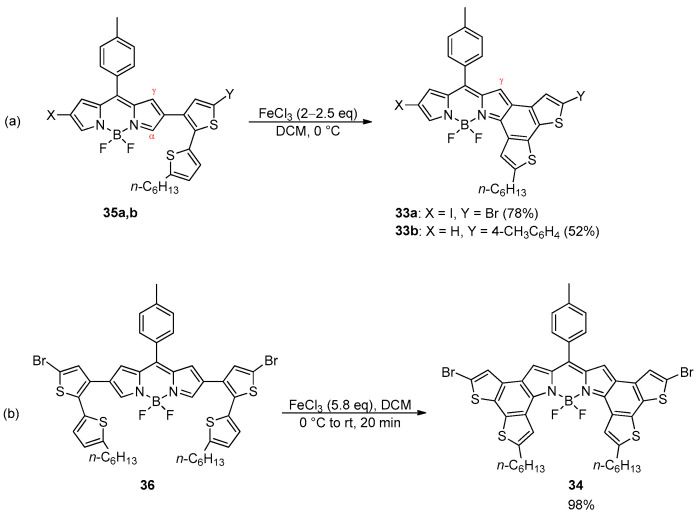 Figure 35
Figure 35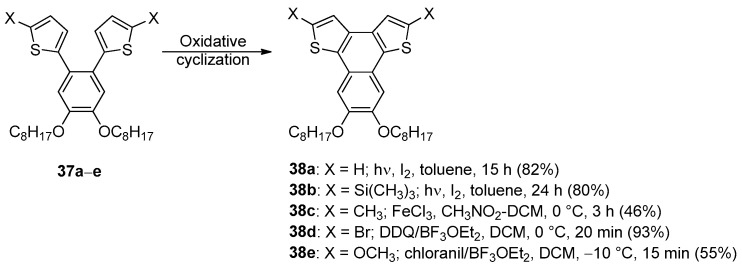 Figure 36
Figure 36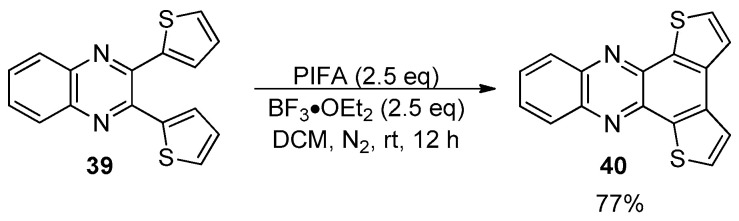 Figure 37
Figure 37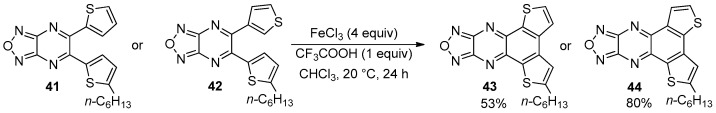 Figure 38
Figure 38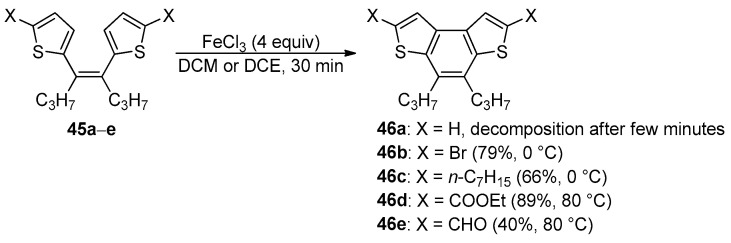 Figure 39
Figure 39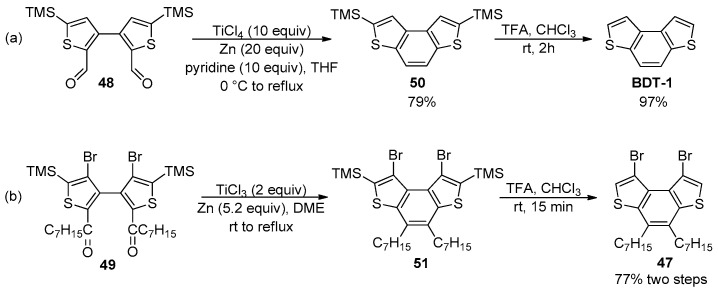 Figure 40
Figure 40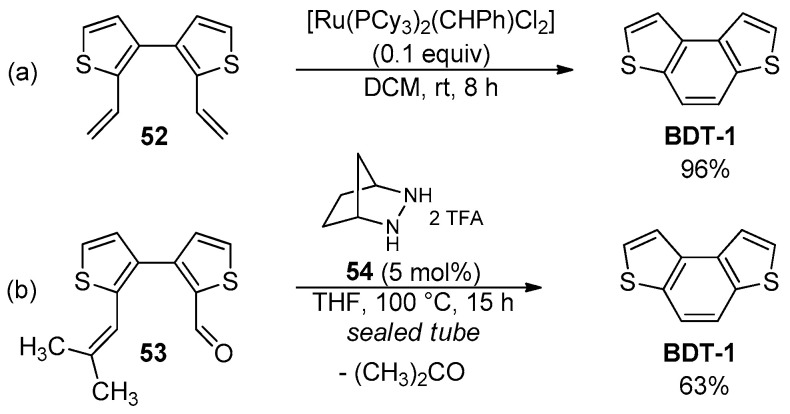 Figure 41
Figure 41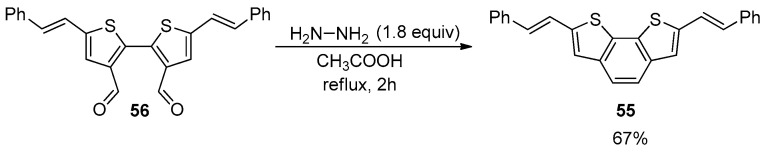 Figure 42
Figure 42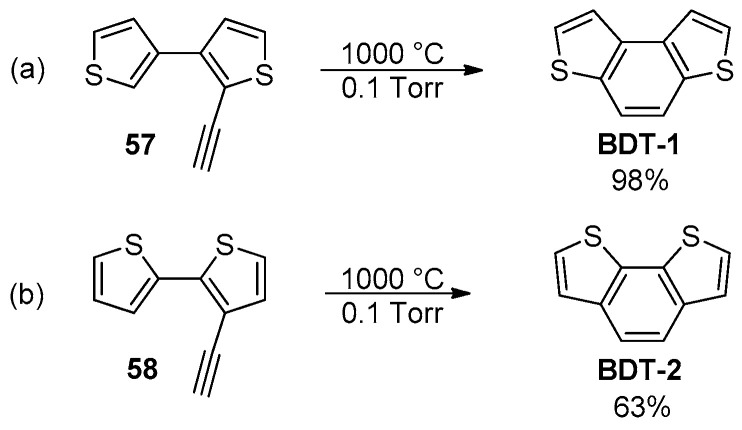 Figure 43
Figure 43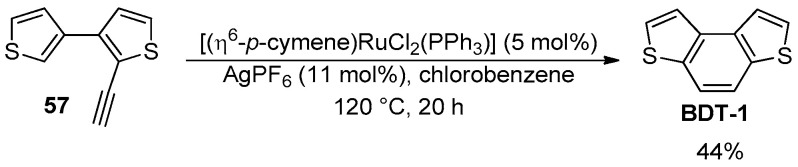 Figure 44
Figure 44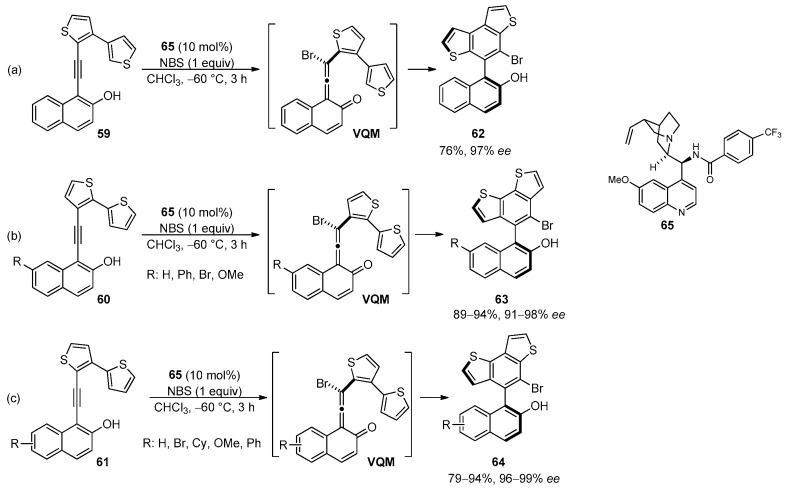 Figure 45
Figure 45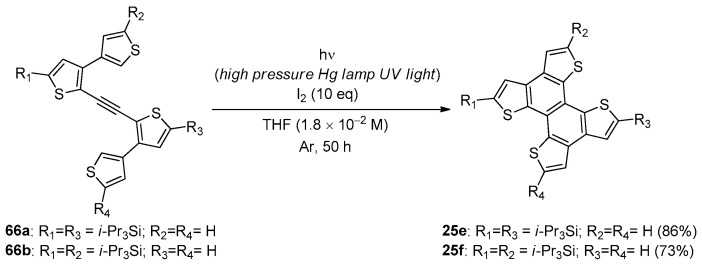 Figure 46
Figure 46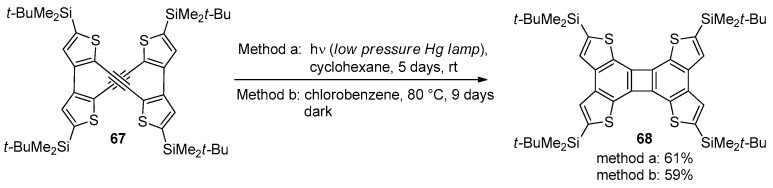 Figure 47
Figure 47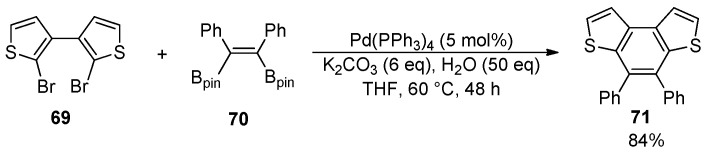 Figure 48
Figure 48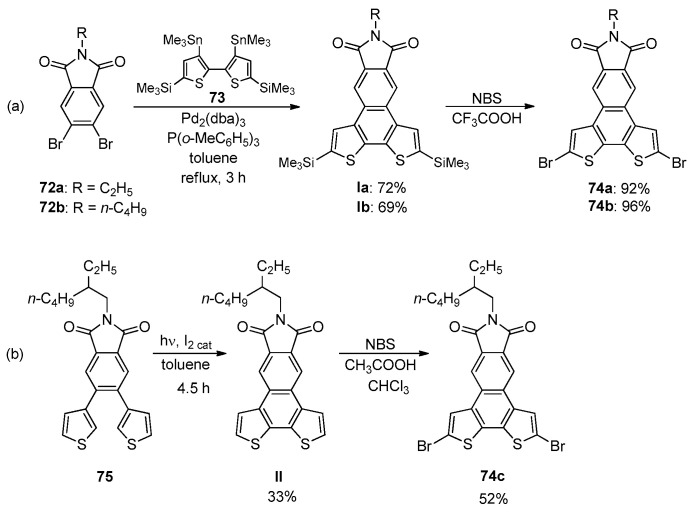 Figure 49
Figure 49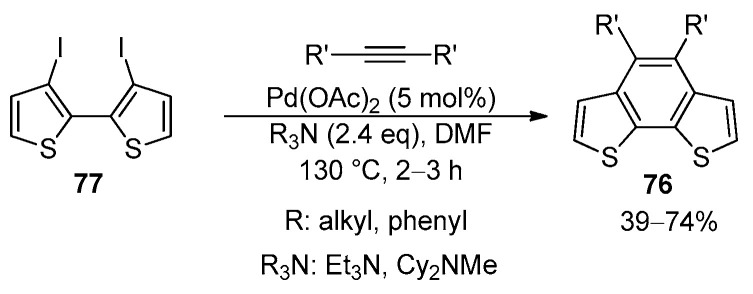 Figure 50
Figure 50Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganic Electronics and Photovoltaics · Synthesis and Properties of Aromatic Compounds · Organic and Molecular Conductors Research
1. Introduction
Chalcogenophenes are five-membered aromatic rings incorporating a chalcogen atom (O, S and Se), known respectively as furan, thiophene and selenophene, which represent fundamental building blocks for the design of functional organic small molecules or polymer-based materials for optoelectronic applications [1,2]. Because chalcogens differ in size, polarizability, and electronegativity [3], their substitution within the chalcogenophene ring can be strategically employed to modulate photophysical properties, aromaticity, reactivity, and redox behavior of chalcogenophene-fused π-conjugated systems. The larger atomic radius and high polarizability of selenium enhance lone-pair participation in intermolecular interactions, improving charge-transport properties [4], while the smaller van der Waals radius and the higher electronegativity of oxygen promote dense packing structure and efficient carrier transport in the solid state [5]. Furthermore, furan derivatives combine strong fluorescence and high dipole moments, enabling superior solubility in polar solvents, unlike sulfur and selenium analogs whose weak fluorescence is generally attributed to the heavy-atom effect [6,7]. On the other hand, aromaticity significantly influences stability and reactivity, generally following the trend benzene > thiophene > selenophene > furan [3], although establishing a definitive ranking remains challenging [8,9]. These chalcogenophenes, being electron-rich, undergo electrophilic substitution faster than benzene, preferentially at the alpha-position (furan > selenophene > thiophene) [10,11], a trend opposite to their aromaticity due to differences in electron delocalization and orbital overlap [8,10]. Moreover, sulfur and selenium can access higher oxidation states through the involvement of d-orbitals, enabling structural modifications such as S,S-dioxides that profoundly alter electronic properties.
The incorporation of chalcogenophenes into extended π-conjugated systems significantly affects molecular planarity, electron distribution, and intermolecular interactions. The planar structure of these compounds, which favors π–π stacking, combined with dense molecular packing and efficient charge-carrier mobility, makes them highly promising building blocks for organic electronic devices. Among these, benzodichalcogenophenes, such as benzodifurans, benzodithiophenes, and benzodiselenophenes, represent a family of tricyclic heteroaromatic compounds formed by fusing two chalcogenophene units onto a benzene ring. These systems have attracted considerable attention over the past decades for applications in organic field-effect transistors (OFETs), organic light-emitting diodes (OLEDs), and photovoltaic cells, owing to their versatile synthesis, chemical stability, and superior solubility compared to polycyclic aromatic hydrocarbon (PAH) analogs. Benzodichalcogenophenes exist as structural isomers depending on the orientation of the fused heteroaromatic rings, enabling property tuning. While structural parameters like bond length and molecular volume remain similar, their chemo-physical properties and intermolecular electronic interactions are generally different. These structural isomers are mainly divided into two linear isomers resembling the anthracene framework (Figure 1), and three angular isomers that are isoelectronic with phenanthrene and are often referred to phenanthrene-like benzodichalcogens (BDF, BDT and BDS, Figure 1). Despite the extensive investigation and review of linear anthracene-like isomers, especially in the context of organic electronic applications [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28], to the best of our knowledge, a comprehensive discussion addressing the synthesis and major applications of phenanthrene-like counterparts BDT, BDF and BDS is still lacking.
This review aims to provide an overview of advances up to the end of March 2025 in the synthesis and applications of BDT, BDF and BDS derivatives. First, this review systematically examines synthetic strategies for constructing the tricyclic core of benzodithiophenes (i.e., benzo[1,2-b:4,3-b′]dithiophene (BDT-1), benzo[2,1-b:3,4-b′]dithiophene (BDT-2) and benzo[1,2-b:3,4-b′]dithiophene (BDT-3)), benzodifurans (i.e., benzo[1,2-b:4,3-b′]difuran (BDF-1), benzo[2,1-b:3,4-b′]dithiophene (BDF-2) and benzo[1,2-b:3,4-b′]dithiophene (BDF-3)), and benzodiselenophenes (i.e., benzo[1,2-b:4,3-b′]bis(selenophene) BDS-1 and benzo[2,1-b:3,4-b′]bis(selenophene) BDS-2). Second, the electrochemical properties of BDT, BDF and BDS are discussed, focusing on the thermodynamic and kinetic factors that differentiate these chalcogen-fused isomers. Third, the most significant applications of these systems in optoelectronics, supramolecular chemistry, and emerging bio-related fields are reported. On the contrary, this review does not cover data reported in the patent literature or those concerning the synthesis and applications of benzotrichalcogenophenes and dione analogs (e.g., benzodifuranones and benzodithiophenone).
2. Synthesis of Benzodichalcogenophenes BDT, BDF and BDS
The distinct electronic properties of furan, thiophene and selenophene, together with their unique chemical behavior within the BDF, BDT and BDS frameworks, have driven the development of a variety of synthetic methodologies aimed at the construction of the tricyclic core. For the sake of clarity, this paragraph is organized into three parts, each addressing a specific heteroatom: beginning with BDT derivatives, for which the literature provides numerous examples, followed by an overview of the synthetic procedures reported for BDFs, and concluding with the limited procedures described to date for BDS systems. The discussion focuses primarily on the cyclization step leading to the formation of the tricyclic skeleton, while the preparation of the corresponding precursors is only briefly mentioned.
2.1. Synthesis of Benzodithiophene (BDT) Scaffolds
The synthesis of BDT-1, BDT-2 and BDT-3 skeleton mainly relies on the construction of the central benzene ring through three different approaches: (i) intramolecular cyclodehydrogenation of 1,2-dithienyl ethenes; (ii) intra and intermolecular annellation reactions of bithienyl derivatives; (iii) domino and/or multicomponent reactions. An alternative synthetic strategy for accessing BDT-2 and BDT-3 scaffolds was also proposed, based on the construction of both thiophene rings from a properly functionalized benzene core.
2.1.1. Intramolecular Cyclodehydrogenation of 1,2-Dithienyl Ethenes
The intramolecular cyclodehydrogenation (ICD) of trans and/or cis isomers of 1,2-di(2-thienyl)ethenes, 1,2-di(3-thienyl)ethenes and 1,2-di(2,3′-thienyl)ethenes represented one of the first practical synthetic approaches developed for the formation of BDT-1, BDT-2 and BDT-3 skeletons, respectively. Currently, the ICD is still a very popular strategy especially to prepare BDT-1 and BDT-2 derivatives, due to the significant improvements achieved in the optimization of the experimental conditions along with the easy and on multigram scale syntheses of dithienyl ethenes. These latter can be generally synthesized through cheap and robust olefination procedures such as the reductive McMurry homocoupling of thienyl carboxaldehydes or ketones, especially for the preparation of symmetrical BDTs, and the Witting reaction and its variants to have access to symmetrical and/or unsymmetrical BDTs.
The ICD strategies mainly used for the synthesis of BDTs include the photochemically induced ICD, also known as the Mallory-type reactions, and the ICD promoted by chemical oxidants under acidic conditions through Scholl-type reactions.
Photochemically Induced ICD Through Mallory-Type Reaction
The Mallory reaction is the photochemical cyclization of stilbene-like derivatives in the presence of catalytic amounts of iodine and is one of the most common methodologies for the synthesis of phenanthrenes, phenacenes, helicenes and other polycyclic (hetero)aromatic compounds [29,30]. This reaction proceeds through a rapid cis/trans photoisomerization of the stilbene, followed by the photo-induced cyclisation of the only cis-isomer and the subsequent iodine-assisted oxidation of the unstable dihydrophenanthrene intermediate in the presence of air. Thus, a cis/trans mixture of diaryl ethenes can be also used in this type of dehydrocyclization, although the yield and the reaction time are generally affected by the cis/trans ratio. The seminal work of Winberg in 1967 [31] demonstrated that the Mallory reaction could be successfully applied for the synthesis of the parent BDT-1, which was obtained in 90% yield by the irradiation of a solution of (E)-1,2-di(2-thienyl)ethene (1) in benzene with a high pressure mercury lamp in the presence of iodine under air for 3 h (Scheme 1a). The same reaction conditions were not as suitable for the photocyclization of ethenes 2 and 3, indeed, while BDT-3 was isolated in moderate yield (47%), the BDT-2 scaffold was not obtained at all, and a complex mixture arising from polymerization processes was observed.
These results were rationalized based on the different behavior of the dihydro intermediates 4–6 (Figure 2) in the oxidation step, in which the abstraction of one hydrogen atom is followed by a second hydrogen abstraction or β-elimination reaction.
While this process was favorable for 4, in case of 5, the abstraction of H″ presumably resulted in the elimination of sulfur and formation of vinylthiyl radicals that in turn produced complex polymer species. For intermediate 6, the possibility to remove H′ and H″ was the same, and then a comparable amount of BDT-3 (47%) and polymer species were formed [32]. Recently, the photochemical cyclization towards the BDT-2 scaffold could be efficiently accomplished starting from the 2,2′-dithienyl derivative 7, from which the naphtho[2,1-b:3,4-b′]dithiophene 8 was isolated in 91% yield (Scheme 2) [33].
On the other hand, BDT-1 systems functionalized with carboxylic acid [34], ester [35], phenyl [36], α-naphthyl [37] and formyl [38] groups were synthesized through oxidative photocyclizations of the corresponding dithienyl ethenes. Mallory’s conditions also afforded the BDT-1 scaffold fused into polyaromatic hydrocarbons [39,40,41,42].
Bromine atoms in the alpha or beta positions of the terminal thiophene rings were also well tolerated under classical Mallory conditions, and bromide 9 [43] and dibromide 10 [44] were efficiently synthesized by photocyclization of dithienyl ethenes 11 and 12 (Scheme 3).
Mallory’s conditions were also effective in promoting the formation of BDT-1 and BDT-2 cores fused with nitrogen-containing heteroaromatic systems [45]. The photocyclization of pyrazines 13 [46] and 14 [47] gave the corresponding quinoxalines 15 and 16 in moderate yields (Scheme 4).
More recently, the photochemical cyclization of 4-methyl-6,7-dithienylcoumarins 17 and 18 provided the corresponding BDT-1- and BDT-2-fused benzo[g]coumarins 19 and 20 in excellent yields (Scheme 5) [48].
The photochemical processes towards functionalized BDT-1 and BDT-3 were also performed in the presence of propylene oxide [49] or methyloxirane [50] as scavenger for hydrogen iodide according to Katz’s conditions [51]. These conditions also allowed photocyclizations in the presence of different heteroaromatic compounds such as 4,5-dithienyl pyrimidines 21a,b and 22a,b, which gave the corresponding BDT-1 and BDT-3-fused systems 23a,b and 24a,b in moderate yields (Scheme 6) [49].
The formation of a BDT-1 core into tetrathienonaphthalenes 25 through the double photocyclization of the corresponding tetra(thien-2yl)ethenes 26 was also deeply investigated, due to their promising properties as p-type organic semiconductors [52] and for their aggregation-induced emission behavior [50]. Besides the first studies reported by Harrit in 1996 [42], alkylated tetrathienonaphthalenes 25a–c were synthesized in moderate to good yields in the presence of p-chloranil as the electron acceptor in both batch and under continuous microflow conditions (Scheme 7) [52].
Regardless of the length of the alkyl chain, these photo-induced cyclization reactions provided products 25a–c in higher yields under microflow conditions than those obtained using traditional batch reactors. The “batch” conditions also provided tetrathienonaphthalene 25d in 56% yield, which was comparable with that obtained under the Mallory–-Katz conditions (64% [50]).
Alternatively, compound 25d was also isolated in comparable yield (61%) by the cobaloxime-catalyzed dehydrogenative photocyclization of 26d (Scheme 8a) [53]. This method represents an advantageous alternative to Mallory–Katz and Scholl reaction (vide infra) since it avoids the use of oxidants and strong acids. Moreover, this approach allowed the synthesis of naphtho[1,2-b:4,3-b′]dithiophene 27 from 1,2-dithienyl benzene (28) in excellent yield (Scheme 8b).
Oxidative Cyclodehydrogenation Through Scholl-Type Reaction
Cyclodehydrogenation promoted by organic or inorganic oxidizing agents under acidic conditions represents a complementary and effective strategy towards the formation of the BDT-1 and BDT-2 skeleton. At the beginning of 2000s, Tovar and Swager [54,55] reported an in-depth study on the chemical and electrochemical oxidation of 1,2-bis-(2-thienyl) and 1,2-bis-(3-thienyl)benzene derivatives, and in particular the FeCl_3_-mediated oxidative cyclization of 1,2-bis-(3-thienyl)benzene derivative 29 provided the corresponding naphtho[2,1-b:3,4-b′]dithiophene 30 in high yield and selectivity (Scheme 9a) [54].
A large molar excess of the oxidant allowed the complete conversion of the starting materials, and any significant polymerization by-products were observed. Otherwise, the formation of the carbon–carbon bond at the β-positions of 1,2-bis-(2-thienyl)benzene 31 to obtain the naphtho[1,2-b:4,3-b′]dithiophene 32 required the protection of the α-positions of thiophene rings to form the desired BDT-1 framework and prevented polymerization processes due to the high spin density of the thiophene radical cation at the 2-position (Scheme 9b).
After this study, the Scholl-type reaction [56] was extensively investigated for intramolecular oxidative C–C bond formation between two thienyl rings to yield BDT-based π-conjugated materials. The latter make use of FeCl_3_ or MoCl_5_ in dichloromethane or organic oxidants, such as dichlorodicyano-p-benzoquinone (DDQ), chloranil or bis(trifluoroacetoxy)iodobenzene (PIFA), in combination with strong Lewis or Brønsted acids. Although the mechanism of Scholl’s reaction is still controversial, the arenium cation and the radical cation mechanism are the most accepted, and both require that the two thienyl rings rely on the same side to form the biaryl linkage. Thus, only cis-1,2-dithienyl ethenes or 1,2-dithienyl benzene derivatives are suitable substrates for this reaction, and they can be prepared through stereoselective olefination reactions (i.e., McMurry coupling of ketones) or palladium-catalyzed cross coupling reactions (i.e., Stille and Suzuki coupling) between aryl halides and thiophene-based organostannane or organoboron compounds. On the other hand, the FeCl_3_-mediated cyclodehydrogenation was found to be a more effective strategy for the construction of the BDT-2 skeleton in comparison with the photochemical approach, and allowed for obtaining the BDT-2 core incorporated into naphthodithiophenes [57], tetrathienoanthracenes [58], dithieneonaphthothiadiazoles [59,60], porphyrins [61], and anthracene-, pyrene- and perylene-base systems [62].
Notably, the formation of the BDT-2 core to yield the corresponding α-fused BODIPY dyes 33a,b and 34 was achieved through the selective oxidative coupling of dithienyl derivatives 35a,b and 36 in the presence of anhydrous FeCl_3_ (Scheme 10) [63].
The α-position of the BODIPY dyes was found to be the most reactive site toward the intramolecular oxidative reaction, providing the γ-fused forms as the main products.
The formation of the BDT-1 scaffold could also be easily accomplished by the oxidative FeCl_3_-, MoCl_5_- or DDQ-mediated cyclodehydrogenation of 1,2-dithienyl benzenes or more complex systems to synthesize naphthodithiophenes [64,65,66], dithieneonaphthothiadiazole [60], tetrathienoanthracenes [67,68,69], pyrene- [40,70] chrysene- [71] and triphenylene-based systems [72], benzo[k]fluoranthene derivatives [73], tetracene diimide [74], functionalized imides [75,76], benzoquinones [77], extended tetracyano-p-quinodimethane [78], highly π-conjugated quinacridones [79], and tetrathienodiborapentacenes [80].
In 2013, a comprehensive study on the intramolecular cyclodehydrogenation of functionalized 1,2-bis(octyloxy)-4,5-bis(2-thienyl)benzenes 37 towards the synthesis of the corresponding naphthothiophenes 38 showed how the nature of the substituents in the alfa positions of the thiophene rings in combination with the nature of the chemical oxidant affected the outcome of the oxidative cyclization (Scheme 11) [66].
Unsurprisingly, the selective intramolecular cyclization of substrates 37a,b could be achieved only under oxidative photochemical conditions, since the use of FeCl_3_ in nitromethane or DDQ-BF_3_•OEt_2_ at 0 °C gave polymerization and deprotection followed by polymerization, respectively, after 20 min.
Although the Scholl reaction has been predominantly applied to electron-donating systems, examples involving electron-withdrawing heterocycles, while less common, are also reported [81,82,83]. The PIFA-BF_3_•OEt_2_-mediated oxidative cyclodehydrogenation of dithienyl quinoxaline 39 provided the corresponding phenazine 40 in good yield in the presence of the free alfa positions of the thiophene rings (Scheme 12) [82].
More recently, the formation of BDT-1 and BDT-3 skeletons was promoted by the FeCl_3_-mediated oxidative cyclodehydrogenation of ortho-dithienyl substituted furazanopyrazines 41 and 42, from which the corresponding polycyclic systems 43 and 44 were isolated in moderate yields (Scheme 13) [83].
While the oxidative cyclodehydrogenation was mainly applied on 1,2-dithienyl benzene derivatives, only one example of cyclization of cis-1,2-dithienyl ethenes was described [84]. The FeCl_3_-mediated cyclization of (Z)-dithienyl ethenes 45 was reported for the synthesis of functionalized 4,5-dipropylbenzodithiophenes 46 (Scheme 14) [84].
The nature of the substituents in the alfa positions and the reaction temperature significantly affected the efficacy and selectivity of this reaction: (i) the complete decomposition of the alkene without α-substituents 45a was observed at room temperature or 0 °C; (ii) the cyclization of dibromo and dialkyl alkenes 45b,c was favored by low temperatures; (iii) the alkenes bearing electron-withdrawing groups 45d,e required higher temperatures (up to 80 °C) for the formation of the corresponding BDT-1 derivatives [84].
2.1.2. Intramolecular and Intermolecular Annellation Reactions of Bithienyl Derivatives
Since the first decade of the 2000s, an alternative approach to the cyclodehydrogenation reactions of dithienyl alkenes or benzene derivatives was explored and applied for the synthesis of BDT-1, BDT-2 and BDT-3, and it involves the construction of their central benzene ring via intra- and intermolecular annellation reactions of the corresponding functionalized 3,3′- and 2,2′-bithienyl compounds.
Intramolecular Annellations
The 3,3′-bithienyl derivatives, properly modified in the alpha positions with carbonyl and/or alkenyl pendants, are readily available by common homo-coupling reactions, and they represent key intermediates to forge the benzene ring of the BDT-1 derivatives through intramolecular McMurry coupling [85,86], the ring-closing metathesis (RCM) [87], and the ring-closing carbonyl–olefin metathesis (RCCOM) [88].
The synthesis of the parent BDT-1 and the dibromide 47 could be achieved by a two-step procedure involving the intramolecular McMurry coupling of dialdehyde 48 [85] and diketone 49 [86], respectively, followed by the smooth and efficient removal of both trimethylsilyl (TMS) groups from intermediates 50 and 51 under acidic conditions (Scheme 15).
On the other hand, the RCM reactions of 2,2′-divinylbiphenyl derivatives have been extensively used to prepare phenanthrene-like compounds, including the parent BDT-1, which was obtained in high yield by the Ru-catalyzed RCM of 2,2′-divinyl-3,3′-bithiophene 52 using the 1st generation Grubbs catalyst (Scheme 16a) [87].
The RCCOM reaction has also proven suitable for the synthesis of a variety of polycyclic heteroaromatic molecules [89], and a metal-free RCCOM protocol [88] promoted by strained hydrazines as organocatalysts was recently developed to prepare functionalized benzo[h]isoquinolines, naphthofurans and thiophene-containing systems such as the parent BDT-1. This latter was isolated in 63% yield by the condensation of the aldehyde 53 in the presence of dialkylhydrazine salt 54 in THF at 100 °C, obtaining acetone as the easily removable waste product (Scheme 16b).
The intramolecular reductive coupling of a 2,2′-bithienyl derivative bearing two formyl groups in the beta positions for the formation of the BDT-2 skeleton was also reported (Scheme 17) [90].
Product 55 was isolated in 67% yield by the hydrazine-mediated cyclization of dialdehyde 56 in acetic acid at reflux, according to the standard Bacon’s conditions [91]. It should be noted that the reductive coupling from bis-tosylhydrazones of 3,3′-bithienyls towards the formation of BDT-1 derivatives was also explored according to Jung’s conditions [92], though low yields were achieved (32–37%) [87].
2-Ethynylbithienyl systems are equally useful and easily accessible intermediates, from which the BDT skeleton can be formed through thermal-induced cyclizations as well as cycloisomerization reactions triggered by electrophilic reagents such as Lewis acids or metal complexes. First studies on the conversion of 2-ethynyl-3,3′-bithiophene (57) and 3-ethynyl-2,2′-bithiophene (58) into the parent BDT-1 and BDT-2, respectively, were reported using the flash vacuum pyrolysis (FVP) (Scheme 18) [93].
Benzodithiophenes were isolated in good to excellent yields (63–98%) and with complete regioselectivity for BDT-1, although this approach has not had further synthetic applications in this context. Otherwise, since the seminal work of Fürstner and co-workers [94], the intramolecular hydroarylation of 2-alkynylbiaryls mediated by metal catalysts or Lewis acid organocatalysts under much milder conditions has been applied for the synthesis of a great variety of phenanthrene-like derivatives and larger poly(hetero)aromatic molecules [95]. This approach was also applied for the synthesis of the BDT-1, which was isolated in moderate yield by the Ru-catalyzed cycloisomerization of 2-ethynyl-3,3′-bithiophene (57) (Scheme 19) [96].
More recently, properly designed 3,3′-, 2,2′- and 3,2′-bithienyls 59, 60 and 61 were employed for the efficient and atroposelective synthesis of axially chiral naphthyl-based BDT derivatives 62, 63 and 64 (Scheme 20) [97].
The reaction proceeded through an intramolecular 6π-electrocyclization of the vinylidene ortho-quinone methide (VQM) intermediate with the thiophene ring, in the presence of N-bromosuccinimide (NBS) as brominating electrophile and the quinine-derived amide 65 as chiral Brønsted base organocatalyst. This procedure was also successfully applied for the synthesis of axially chiral naphthyl-based BDF-1 derivative (90% yield, 93% ee) and thieno[3,2-e]benzofurans (up to 97% yield, 96% ee) [97].
The chemistry of the intramolecular annellation of alkynylated biaryls was also applied by Yamaguchi et al. [98] for the effective one-pot synthesis of tetrathienonaphthalenes 25e,f, which were obtained by irradiation of a THF solution of bis(bithienyl)acetylenes 66a,b with a high-pressure mercury lamp in the presence of a large molar excess of I_2_ (Scheme 21).
The formation of the corresponding 6-endo cyclized products, 25e,f, presumably occurred through a one-pot process that involved an iodine-promoted electrophilic monocyclization, followed by a second UV-induced electrocyclization and aromatization. The same authors described the synthesis and the intriguing reactivity of the thiophene-containing bisdehydro[12]annulene 67, which was able to undergo a metal-free [2+2]-type alkyne cycloaddition under either photoirradiation or mild thermal conditions (Scheme 22) [99].
The possibility of obtaining the BDT-based cycloadduct 68 with mild heating is likely ascribed to the less aromatic character of thiophene, which was more prone to losing aromaticity during the thermal process.
Intermolecular Annellations
A typical example of intermolecular annellation for building polycyclic (hetero)aromatic compounds makes use of palladium-catalyzed cross-coupling reactions, including the Suzuki and the Stille coupling, which were also employed for the formation of the central benzene ring of the BDT-1 and BDT-2 skeleton. The double Pd(0)-catalyzed Suzuki coupling between dibromide 69 and the (Z)-1,2-bis(pinacolatoboryl)stilbene 70 provided the disubstituted BDT-1 derivative 71 in 84% yield (Scheme 23) [100].
Compound 71 was also obtained in 70% yield through the photocyclization of 1,2-diphenyl-1,2-di(thiophene-2-yl)ethene under classical Mallory conditions [36]. The double Pd(0)-catalyzed Stille coupling between the dibromobenzene imides 72a,b and 2,2-bithienyl distannane 73, followed by the one-pot process involving the removal of the TMS groups and bromination, gave the BDT-2 bromides 74a,b in good yields (Scheme 24a) [101].
This two-step procedure for the synthesis of bromides 74 was more efficient than that of the construction of the BDT-2 accomplished by Mallory-type photocyclization of 75 (Scheme 24b) [102].
The synthesis of 4,5-disubstituted BDT-2 derivatives 76 was also reported via the intermolecular Pd(OAc)2-catalyzed annulation of 3,3′-diiodo-2,2′-bithiophene (77) with internal alkynes in the presence of a trialkyl amine in DMF at 100–130 °C under ligandless conditions (Scheme 25) [103,104].
Besides the palladium chemistry, the Rh(III)-catalyzed dehydrogenative coupling of heterobiaryls with alkynes represents a convenient alternative towards the synthesis of fused polycyclic heteroarenes [105]. This approach does not require a pre-functionalization of the biaryl systems since it generally proceeds via a direct C–H bond cleavage followed by the alkyne insertion and reductive elimination. Until now, attempts to obtain the BDT skeleton through this approach have not provided satisfactory results, such as the rhodium-catalyzed annulation of 2,2′-bithiophene (78) with diphenylacetylene, from which a mixture of 4,5-diphenyl substituted BDT systems 71 and 79 was obtained in very low yield (Scheme 26) [106].
More recently, different BDT-fused isoquinolines 80–82 were obtained in good to excellent yield by the iridium-catalyzed [2+2+2] cycloaddition of nitriles with bithiophenes 83–85 bearing two alkynyl pendants (Scheme 27) [107].
The metal-catalyzed [2+2+2] cycloaddition reaction is indeed an atom-economical approach for the formation of several carbon–carbon and carbon–heteroatom bonds in one step and allows for straightforward access to complex polycyclic (hetero)aromatic compounds [108]. The scope of this reaction was deeply investigated, demonstrating the high versatility and efficacy of this approach, especially towards isoquinolines 80 and 81.
2.1.3. Domino and Multicomponent Reactions
The atom-economical domino and multicomponent reactions have proven to be a convenient and alternative route to access BDT derivatives. In this context, the regioselective construction of the BDT-2 scaffold could be achieved by a domino one-pot protocol involving the direct arylation reaction and a cross-aldol intramolecular condensation starting from readily available reagents 86 and 87 (Scheme 28) [109,110].
The BDT-2 derivative 88a was isolated in high yield, and the presence of the electron-withdrawing carboxylate ester was found to be essential in favoring the intramolecular cross-aldol condensation [109]. This protocol was also applied for the synthesis on a gram-scale of a BDT-2 derivative 88b bearing a long alkyl chain to improve its solubility in common organic solvents and to then facilitate its usage in organic devices [110].
A complementary strategy to build the parent BDT-2 made use of a two-step reaction involving the alkylation at low temperature of 2-bromo-3-(phenylsulfonylmethyl)thiophene (89) with 3-bromomethylthiophene (90), followed by the intramolecular direct arylation of sulfone 91 and the concomitant elimination of phenylsulfinic acid to yield the aromatic structure (Scheme 29) [111].
An efficient and highly regioselective multicomponent approach towards π-extended (hetero)aromatic compounds involves the use of norbornadiene (NBD) as a key building block in the palladium-catalyzed C–H functionalization of (hetero)aryl halides, where the NBD derivatives act as an ortho-C–H activator and ethylene synthon [112]. As far as the BDT skeleton, the palladium-catalyzed three-component reaction between the easily available thienyl derivatives 92 and 93 and NBD provided the substituted BDT-1 94 in good yield (Scheme 30).
DFT calculations suggested that the reaction occurred via the decarboxylative NBD-mediated palladium cascade, in which the Pd(II)-Pd(IV) oxidative addition seemed to be the rate-determining step, followed by the retro-Diels-Alder (rDA) reaction of the NBD-fused intermediate to yield the phenanthrene-like BDT-1 scaffold.
The palladium-catalyzed multicomponent coupling reaction of bromothiophenes 95 and NBD, followed by rDA via a 2:1 annulation, was also employed to build the BDT-2 and BDT-3 frameworks (Scheme 31) [113].
Tetrabutylammonium iodide (TBAI) significantly affected the selectivity of this reaction towards the cis- and trans-annulation, though for both BDT derivatives, low yields were achieved.
Finally, a one-pot process involving the N-methyl-3-(2-thienyl)-N-(2-thienylsulfonyl)propiolamide 98 was reported for the synthesis of the parent BDT-1 (Scheme 32) [114].
The base-assisted photoinduced reaction of a toluene solution of 98 afforded BDT-1 in 57% yield via the one-pot Smiles rearrangement and Mallory-type oxidative cyclization, followed by the removal of the sulfonylamide moiety. This latter step was proven to occur through a ring-opening reaction followed by elimination of an isocyanic intermediate and its trapping by morpholine to yield morpholine-4-carboxamide 99.
2.1.4. Construction of Both Thiophene Rings from Arylethynylated Naphthalenes
The metal-free thienannulation of arylethynylated naphthalenes 100a,b and 101, via ortho-C-H bond cleavage promoted by elemental sulfur in DMF at 140 °C, provided the corresponding naphtho[2,1-b:3,4-b′]dithiophene 102a,b and naphtho[1,2-b:3,4-b′]dithiophene 103 in moderate yields (Scheme 33a,b) [115,116].
In addition, the reaction of 2-fluoroethynylbenzenes 104 with Na_2_S as nucleophile efficiently led to the formation of the BDT-2 skeleton in dithienobenzothiadiazoles 105 via a cascade sequence involving nucleophilic aromatic substitution followed by anionic cyclization onto the pendant alkyne (Scheme 33c) [117]. Remarkably, this thiolation annulation proceeds without any transition-metal catalyst and in excellent yields within 10 min.
2.2. Synthesis of Benzodifuran (BDF) Scaffolds
The earliest approach to the preparation of parent BDF-1 involved constructing the central benzene ring via the oxidative photochemical cyclization of difurylethene [31]. Although this method is widely used for the synthesis of BDT cores, its use for BDF has remained limited. Notably, in 2017, the formation of the central benzene ring of BDF-1 skeleton in derivatives 106 [118] and 107 [119] was accomplished by photoinduced oxidative cyclization in a polar protic solvent (EtOH), without the need for any oxidant and catalyst (Scheme 34a,b).
BDF-1 derivatives 106 were obtained in moderate yields by the direct intramolecular annulation of 2,3-di(fur-2-yl)arylchromen-4-ones 108 in a mixture of water and EtOH at room temperature (Scheme 34a), while the photocyclization of 109 under similar reaction conditions provided furan[3,2-e]benzofurans 107 in 42–65% yields by a photoinduced process involving keto-enol tautomerization steps (Scheme 34b). These synthetic methodologies also enabled the preparation of related tricyclic analogs, such as thieno[3,2-e]benzofuran 110 and 111 (Scheme 34c,d), which were obtained through the photocyclization of 112 and 113, respectively, in yields comparable to those of the corresponding BDF-1 derivatives 106 and 107.
Aside from these examples, the formation of BDF-1, BDF-2 and BDF-3 cores mainly relies on three strategies: (i) construction of both furan rings starting from functionalized benzenes; (ii) formation of a furan ring from suitably modified benzofurans; (iii) formation of a benzofuran core from furan-bridged enynes.
2.2.1. Construction of Both Furan Rings from Functionalized Benzenes
Properly designed hydroquinones can serve as versatile starting materials for the synthesis of various isomeric benzodifurans. Parents BDF-1, BDF-2 and BDF-3 could be obtained through a Sonogashira coupling between trimethylsilylacetylene and diester of diiodohydroquinone 114, diiodocatechol 115 and diiodoresorcinol 116, respectively, followed by a sequential cyclization and desilylation reactions promoted by tetrabutylammonium fluoride (Scheme 35) [120].
The same approach was also employed for the synthesis of functionalized BDF-3 scaffolds [121]. Alternatively, BDF-1 systems 117a,b could be accessed through one-pot reactions involving the addition of hydroquinone 118 to bromoalkynes 119a,b, followed by the intramolecular cyclization via palladium-catalyzed C–H bond functionalization (Scheme 36) [122].
This methodology [123], which does not require ortho-halogenated phenols, represents a valid alternative to the Sonogashira coupling/cyclization strategy discussed above and provides the final BDF-1 systems in similar yields to those reported in Scheme 35.
More recently, chloroolefins 120, 121 and 122, obtained via selective nucleophilic addition of hydroquinone, resorcinol, and catechol, respectively, to 1-chloro-1-octyne, have proven to be suitable intermediates for the synthesis of the corresponding alkylated BDFs systems 123–125 (Scheme 37) [124].
This transformation proceeds via an intramolecular palladium-catalyzed C–H cyclization carried out using a catalytic system made of Pd(OAc)2 and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos). The cyclization of 120 and 122 proceeded with no complete selectivity due to concurrent dual C–H cyclization within the same molecules. Thus, regioisomers 123 and 126 were isolated as a mixture (75/25 ratio) in 68% yield from chloroolefin 120, while 125 and 127 were obtained as a mixture in a similar ratio and yield from chloroolefin 122 (Scheme 37a,c). The BDF-2 derivative 124 was isolated as a single isomer in 74% yield, presumably because of the limited number of activatable C−H bonds (Scheme 37b).
Substituted hydroquinone 128 was used to prepare BDF-1 derivatives 129a,b in high yields via alkylation and dehydrative cyclization reactions (Scheme 38) [125].
Notably, for the compound 129a, the alkylation and cyclization/dehydration were accomplished in a one-pot process under refluxing acetone. A similar strategy was also employed for the preparation of difuro[2,3-e:2, 3-G]indoles [126]. Conversely, a Cs_2_CO_3_-promoted substitution-elimination process between nitroallylic acetate 131 with catechol (132) gave the corresponding BDF-2 derivative 133 in moderate yield (Scheme 39) [127].
A reasonable mechanism for this transformation involves a Friedel–Crafts-type reaction through an S_N_2′ process, followed by an intramolecular oxa-Michael cyclization and the subsequent energetically favored aromatization of the furan cores.
Besides the methodologies mentioned above, it should be noted that the BDF-2 skeleton can be synthesized by a Brønsted acid-mediated nucleophilic addition-carbocyclic rearrangement cascade reaction between catechol (132) with bis[(trimethylsilyl)oxy]cyclobutene (134) (Scheme 40) [128].
While BDF-2 derivative 135 was isolated in 49% yield, the same reaction using resorcinol gave a mixture of two regioisomers in 40% overall yield. In addition, substituted BDF-2 derivative 136 can be prepared in 61% overall yield by a two-step procedure involving cyclization of 3,6-dialkynylcatechol 137 into a thermally stable organozinc intermediate 138, followed by Negishi coupling with bromide 139 (Scheme 41) [129].
This protocol offers a divergent-oriented synthetic approach, enabling the preparation of a broad range of furan-based polyaromatic compounds [5].
Naphthaquinone (140) was also employed as a starting material for the synthesis of BDF-1 derivatives through two different strategies, both based on a [3+2] cyclization with olefines [130] or propargyl alcohols [131]. The first approach involved a transition-metal-free oxidative C−H transformation of quinones followed by dehydrogenation. Specifically, the [3+2] cyclization of 140 with 1-methyl-4-vinylbenzene (141), followed by oxidative cyclization with an additional olefin molecule, afforded the tetrahydrobenzodifuran 142 (Scheme 42) [130].
Subsequent oxidation of 142 with DDQ provided the benzodifuran 143 in 39% overall yield over two steps. The second strategy involved the formation of the intermediate 144 through an aza-Michael/Michael/annulation sequence starting from ynone 145 and 140 (Scheme 43) [131].
5-Hydroxybenzofuran 144 underwent a Zn-promoted [3+2] cyclization with propargyl alcohol 146 to afford the BDF-1 derivative 147 in 44% overall yield.
Finally, highly functionalized BDF-1 and BDF-2 systems were obtained by the construction of both furan rings from properly functionalized biphenols via an extended Pummerer annulation using substituted ketene dithioacetal monoxides and trifluoroacetic anhydride [132].
2.2.2. Formation of a Furan Ring from Modified Benzofurans
Functionalized benzofuran 148 was successfully employed for the one-pot synthesis of BDF-3 derivatives 149a,b via a sequential addition and intramolecular cyclization reaction of 148 with terminal acetylenes in the presence of PPh_3_ and a solid base nanocatalyst made of KF impregnated on the natural zeolite clinoptilolite (KF/CP) in water at room temperature (Scheme 44) [133].
Conversely, the BDF-3 scaffold can be also formed by an acid-catalyzed rearrangement of 4-acetoxy-9-furylnaphtho[2,3-b]furans 150, from which functionalized naphthodifurans 151 were isolated in 37–55% yield (Scheme 45) [134,135].
2.2.3. Formation of a Benzofuran Core from Furan-Bridged Enynes
A less common approach to the BDF-1 and BDF-3 core involved the coupling of furan-bridged enynes with carbene complexes to obtain the benzofuran core [136]. This strategy takes advantage of the well-known and robust chemistry of Fischer carbene complexes, and in this case relies on the coupling of complex 152 with conjugated dienynes 153 and 154, in which the central alkene was included within the furan ring (Scheme 46a,b).
Both regioisomers 153 and 154 provided the corresponding BDF-1 and BDF-3 derivatives 155 and 156 in very good yields when reacted with 152 at 80 °C in dioxane, followed by the treatment with H_2_SO_4_ at room temperature. The same procedure was also successfully applied for the synthesis of thienobenzofurans 157 and 158, which were isolated in 79 and 84% yield, respectively, starting from enynes 159 and 160 (Scheme 46c,d).
2.3. Synthesis of Benzodiselenophene (BDS) Scaffolds
In contrast to BDF and BDT derivatives, the synthetic methodologies reported so far for the construction of benzodiselenophene core BDS-1 and BDS-2 are still limited. They generally involve the formation of the central benzene ring via (i) the intramolecular McMurry reaction of dialdehyde 161 to give 162 (Scheme 47a) [137], (ii) the double Suzuki coupling between functionalized 2,2′-biselenophene 163 with dioctylbis(pinacolatoboryl)alkene (164) for system 165 (Scheme 47b) [138], (iii) the oxidative photocyclization of dibromide 166 for compound 167 (Scheme 47c) [139].
An alternative pathway towards the BDS-2 skeleton was also reported, and it involves the construction of both selenophene rings [140]. As reported in Scheme 48, this transformation starts with the in situ formation of phenylselenolate by reaction of dibromide 168 with sodium selenide, the latter being obtained by the reduction of selenium powder with NaBH_4_ in ethanol. Then, phenylselenolate species reacted with the triple bonds on the starting material, leading to the construction of both selenophene rings fused to the benzene core. The BDS-2 system 169 was isolated in 80% overall yield.
3. Electrochemical Properties of BDT and BDS Derivatives and Mechanistic Insights
To comprehend the polymerization and redox behavior of BDT and BDS, one must first establish the theoretical underpinnings governing the electrochemistry of fused heterocyclic systems. The electrochemical behavior of these molecules is a direct manifestation of their electronic structure, specifically the energy and distribution of their frontier orbitals.
3.1. Molecular Orbital Theory: Sulfur vs. Selenium
The primary distinction between BDT and BDS lies in the heteroatom. Sulfur, a third-period element, and selenium, a fourth-period element, impart different electronic characteristics to the fused benzo-dichalcogenophene core.
3.1.1. Ionization Potential and Electronegativity
Selenium is less electronegative than sulfur (2.55 vs. 2.58 on the Pauling scale) and possesses a lower ionization potential (9.75 eV for Se vs. 10.36 eV for S). In the context of a conjugated aromatic system, the heavier atom’s valence orbitals (4p for Se vs. 3p for S) are higher in energy and more diffuse. This results in a destabilization of the Highest Occupied Molecular Orbital (HOMO) in selenium-containing heterocycles relative to their sulfur counterparts. A higher HOMO energy level implies that the molecule requires less energy to remove an electron, theoretically leading to a lower electrochemical oxidation potential (Eox).
3.1.2. Polarizability and Orbital Overlap
The larger size of the selenium atom enhances the polarizability of the electron cloud. This increased polarizability facilitates stronger intermolecular orbital overlap between adjacent molecules or polymer chains. In solid-state lattices, this manifests as enhanced transfer integrals, which are the quantum mechanical matrix elements describing the probability of charge hopping between sites. Consequently, BDS derivatives often exhibit higher charge carrier mobilities than BDT analogs, a trend consistently observed in OFET measurements.
3.1.3. Spin-Orbit Coupling
The “heavy atom effect” introduces significant spin-orbit coupling (SOC) in selenium derivatives. While primarily relevant for photophysical transitions (promoting intersystem crossing to triplet states), SOC also influences the electrochemical stability of radical cation intermediates. The stabilization of the radical cation is crucial for the electropolymerization mechanism, as it dictates the lifetime of the reactive species and the selectivity of the coupling reaction.
3.2. The Electric Double Layer and Solvent Effects
Electrochemical reactions occur at the interface between the electrode and the electrolyte solution. The structure of this interface, the electric double layer, governs the kinetics of electron transfer. For hydrophobic organic molecules like BDT and BDS, the solvent choice is critical.
3.2.1. Solvent–Solute Interactions
Common solvents for electropolymerization include acetonitrile (ACN), dichloromethane (DCM), and nitrobenzene. However, BDT and BDS monomers, particularly those with fused ring systems, often suffer from limited solubility in ACN. DCM is preferred for its solvency but has a narrow electrochemical window.
3.2.2. Boron Trifluoride Diethyl Etherate (BFEE)
A significant advancement in the electropolymerization of thiophene-based systems is the use of boron trifluoride diethyl etherate (BFEE) as both solvent and catalyst [141]. BFEE is a Lewis acid that can form complexes with the aromatic rings, lowering the resonance energy and effectively reducing the oxidation potential of the monomer. This allows polymerization to proceed at milder potentials, avoiding the “polythiophene paradox,” where the high potential required to oxidize the monomer leads to the degradation (overoxidation) of the resulting polymer. The interaction between the Lewis acid and sulfur/selenium lone pairs stabilizes the cationic intermediates, promoting the formation of high-quality, linear polymer films with fewer structural defects.
3.3. Mechanisms of Electrochemical Polymerization
The formation of BDT and BDS polymers via electrochemical oxidation is a complex process involving heterogeneous electron transfer and homogeneous chemical coupling [55]. The generally accepted mechanism is the E(CE)n (Electrochemical–Chemical–Electrochemical) pathway, which proceeds through the generation of radical cations [55]. Understanding the precise sequence of bond-forming events is essential for controlling the regioregularity and molecular weight of the polymer.
3.3.1. Stepwise Mechanism of Radical Cation Coupling
The polymerization initiates at the anode surface and propagates through a sequence of oxidation and coupling steps. The fundamental mechanism for BDT (and by extension BDS) can be broken down into four distinct phases: Oxidation, Dimerization, Aromatization, and Propagation.
Phase I: Anodic Oxidation (E)
The neutral monomer (M), diffusing from the bulk solution to the electrode surface, loses an electron to form a radical cation (M^+•^). This process is diffusion-controlled and irreversible.
For BDT and BDS, the highest spin density of the unpaired electron is located at the α-positions (C2 and C7) of the terminal thiophene/selenophene rings. This localization directs the regioselectivity of the subsequent coupling.
Phase II: Radical Copling (C)
Two radical cations couple to form a dicationic σ-dimer (M_2_^2+^). This step is the rate-determining step in the polymerization under conditions of high radical concentration (high current density) [142].
Alternatively, a radical cation may attack a neutral monomer (M^+•^ + M ⟶ (M − M)^+•^), followed by a second oxidation. However, kinetic studies suggest the radical–radical coupling is favored due to the high local concentration of radicals at the electrode interface.
Phase III: Deprotonation and Re-Aromatization (C)
The formation of the σ-bond disrupts the aromaticity of the heterocyclic rings. To restore the stable aromatic system, the dicationic dimer rapidly eliminates two protons (2H^+^).
The release of protons lowers the pH at the electrode interface, which can catalyze side reactions if not managed. The resulting neutral dimer (M_2_) has a more extended conjugation length than the monomer.
Phase IV: Chain Propagation (E)
Because the ionization potential decreases with increasing conjugation length, the dimer (M_2_) is more easily oxidized than the monomer (Eox (M_2_) < Eox (M)). Therefore, at the potential applied to oxidize the monomer, the dimer is immediately oxidized to a radical cation and couples with other radicals (monomers or oligomers), extending the chain [142].
This cascade continues until the oligomer becomes insoluble in the electrolytic medium and precipitates onto the electrode, forming a film.
3.4. The Sigma σ vs. ᴨ Dimer Intermediates
Recent spectroelectrochemical and NMR studies have added nuance to the classical mechanism by identifying specific intermolecular interactions that precede bond formation [142]. This is particularly relevant for planar systems like BDT and BDS.
3.4.1. The ᴨ-Dimer Assembly
Before the formation of a covalent bond, radical cations of planar conjugated systems tend to stack face-to-face to form ᴨ-dimers ([M_2_]^2+^ or [M_2_]^+•^). These aggregates are stabilized by the overlap of the ᴨ-orbitals and dispersion forces. For BDT and BDS, the large, flat aromatic core favors strong ᴨ-dimerization. This pre-organization aligns the molecules in a geometry favorable for α–α coupling, reducing the activation energy for the formation of the σ-bond.
In situ spectroelectrochemistry reveals distinctive absorption bands in the near-infrared (NIR) region attributed to these ᴨ-dimers, which are distinct from the isolated radical cation absorptions.
3.4.2. The σ-Dimer Transition State
The transition from the ᴨ-stacked aggregate to the covalently bonded σ-dimer involves the rehybridization of the α-carbons from sp^2^ to sp^3^. This σ-dimer is a distinct intermediate that has been trapped and characterized in sterically hindered oligothiophenes. In BDT electropolymerization, bulky substituents can sterically encumber this transition, potentially slowing the polymerization rate or necessitating higher overpotentials (Table 1).
3.5. Regioselectivity and Structural Defects
The ideal electropolymerization of BDT/BDS occurs strictly through the α-positions to yield a linear, conjugated backbone. However, the high reactivity of the radical cation can lead to “mislinking” defects.
β-Coupling: Coupling at the β-positions disrupts the conjugation and linearity of the polymer. In fused systems like BDT, the β-positions are part of the fused benzene ring or adjacent to the sulfur, making them sterically and electronically less favorable for coupling than in simple thiophenes. Nevertheless, at high potentials, the selectivity decreases, leading to cross-linking and disordered films [143].
Substituent Blocking: Strategic placement of alkyl or alkoxy chains not only solubilizes the polymer but also sterically directs coupling to the α-positions, enhancing the regioregularity of the electropolymerized film [144].
3.6. Comparative Electrochemical Properties of BDT and BDS Monomers
The substitution of sulfur with selenium induces specific shifts in the redox properties of the monomers. These shifts are quantifiable through cyclic voltammetry (CV) and are critical for energy level matching in device applications.
3.6.1. Oxidation Potentials and HOMO Levels
Experimental data from cyclic voltammetry consistently demonstrates that BDS derivatives are easier to oxidize than their BDT counterparts [145]. The oxidation potential (Eox) serves as a proxy for the HOMO energy level.
3.6.2. Observed Potentials
In a direct comparison of benzo[1,2-b:4,5-b′]dichalcogenophenes, the anodic peak potential (Epa) shifts cathodically (to less positive values) as the chalcogen becomes heavier.
BDT Monomer: Epa ~+0.95 V vs. Fc|Fc^+^.
BDS Monomer: Epa ~+0.89 V vs. Fc|Fc^+^.
BDTe (Tellurium) Monomer: Epa ~+0.48 V vs. Fc|Fc^+^.
This trend corresponds to a destabilization of the HOMO level. Using the standard approximation, , the HOMO levels are estimated as follows:
BDT: −5.60 eV
BDS: −5.50 eV
The 0.1 eV elevation in the HOMO of BDS is attributed to the lower ionization potential of selenium compared to sulfur. This makes BDS a stronger electron donor, which is advantageous for creating low-bandgap polymers when paired with strong acceptors, but it also implies that BDS-based materials may be slightly more susceptible to oxidative degradation in air.
3.6.3. Structural Influence on Redox Behavior
The substituents attached to the BDT/BDS core modulate these intrinsic potentials significantly (Table 2) [146].
3.6.4. Alkoxy vs. Alkyl Side Chains
Alkoxy-BDT: The presence of oxygen atoms attached directly to the benzene core (e.g., di(alkoxy)BDT) raises the HOMO level significantly due to the mesomeric (+M) electron-donating effect of the oxygen lone pairs. This results in a cathodic shift of the oxidation potential by approximately 0.2–0.3 eV compared to alkyl-substituted BDTs. While this facilitates electropolymerization at lower potentials, it can reduce the open-circuit voltage (V_oc_) in solar cells, as Voc is proportional to the difference between the donor HOMO and acceptor LUMO.
Alkyl-BDT: Alkyl chains exert a weak inductive (+I) effect. Consequently, alkyl-BDTs have deeper (more negative) HOMO levels than alkoxy-BDTs. This deeper HOMO is desirable for oxidative stability and high V_oc_, but it requires higher potentials for electropolymerization, increasing the risk of overoxidation.
3.6.5. Two-Dimensional Conjugation
Attaching thiophene or selenophene rings orthogonally to the main chain creates “2D-conjugated” BDTs.
Effect: These side groups extend the conjugation length and absorption cross-section. Electrochemically, a thienyl side group is less electron-donating than an alkoxy group. Therefore, 2D-conjugated BDTs typically exhibit oxidation potentials intermediate between alkyl- and alkoxy-BDTs, offering a balanced trade-off between stability (deep HOMO) and coverage (broad absorption).
3.6.6. Band Gap Considerations
The electrochemical band gap is defined as the difference between the onset of oxidation (HOMO) and the onset of reduction (LUMO). The substitution of S with Se consistently narrows the band gap. While the HOMO is raised, the LUMO levels of BDT and BDS are often comparable or only slightly lowered in BDS. The net result is a reduction in Eg by approximately 0.1 eV.
The optical band gap , determined from the absorption edge, is typically lower than the electrochemical gap due to the exciton binding energy (the energy required to separate the electron–hole pair created by photon absorption). In BDS systems, the higher polarizability of Se can reduce the exciton binding energy, bringing and closer together than in BDT systems.
3.7. Properties of Electropolymerized Films
While chemical coupling (e.g., Stille, Suzuki) is preferred for bulk synthesis, electropolymerization offers unique advantages for BDT and BDS derivatives [55,145,146,147]. The polymerization of BDT and BDS monomers yields conductive films whose properties are distinct from their monomeric precursors. The electropolymerization process essentially “locks” the monomer units into a conjugated backbone, creating a material with delocalized electronic states.
3.7.1. Charge Carrier Mobility and Transport
One of the most compelling advantages of BDS derivatives is their superior charge carrier mobility. This enhancement is a direct consequence of the heavy atom effect and the structural packing it induces.
3.7.2. Interchain Interaction and Transfer Integrals
The larger orbitals of selenium (4p) compared to sulfur (3p) extend further from the atomic nucleus. This extension facilitates greater orbital overlap between adjacent polymer chains in the solid state. Crystallographic and computational studies reveal that BDS polymers often exhibit short interchain Se•••Se distances, significantly shorter than the sum of their Van der Waals radii. These interactions create 3D networks for charge transport, allowing charge carriers (holes) to “hop” between chains more efficiently than in BDT polymers, where Se•••Se interactions are weaker.
In comparative studies of copolymers (e.g., with thienothiophene), BDS-based polymers have demonstrated hole mobilities up to an order of magnitude higher than their BDT analogs (1.35 × 10^−3^ cm^2^ V^−1^ s^−1^ for PBDS vs. lower values for PBDT). In highly ordered ladder-type polymers, BDS incorporation has pushed mobilities exceeding 0.1 cm^2^ V^−1^ s^−1^.
3.7.3. Reorganization Energy
According to Marcus theory, the rate of electron transfer is inversely proportional to the reorganization energy (λ), the energy cost associated with the structural deformation of the molecule upon charging. DFT calculations indicate that BDS derivatives possess lower reorganization energies than BDT derivatives. The rigid, heavy BDS core undergoes less geometric distortion upon oxidation to the radical cation, thereby facilitating faster charge transfer kinetics.
3.7.4. Morphology and Structural Ordering
Electrochemical growth allows for the formation of ordered films, but the degree of order depends on the monomer structure [148].
Both BDT and BDS are planar, but BDS polymers tend to adopt more planar backbone conformations due to non-covalent intramolecular interactions (e.g., Se•••O or Se•••F interactions with side chains or acceptors). This planarity enhances the effective conjugation length. BDT polymers often form amorphous or semi-crystalline films. In contrast, the strong intermolecular interactions in BDS polymers often drive the formation of highly crystalline domains. While this improves mobility, it can also lead to excessive aggregation, which may be detrimental for phase separation in bulk heterojunction solar cells if not carefully controlled [149].
3.7.5. Redox Stability and Doping
The stability of the p-doped state (the oxidized polymer) is crucial for applications like OECTs and sensors.
PBDT: It generally exhibits excellent oxidative stability. The radical cations are stable and reversible. However, “overoxidation” at potentials > 1.2 V (vs. Ag/AgCl) can lead to nucleophilic attack by solvent impurities (e.g., water) on the backbone, breaking conjugation [150,151].
PBDS: Being more electron-rich, PBDS is easier to dope (oxidize). This allows for stable operation at lower voltages, reducing power consumption in devices. However, the higher HOMO level can make the neutral polymer prone to spontaneous oxidation by atmospheric oxygen (unintended doping), which degrades the ON/OFF ratio in transistors. To mitigate this, electron-withdrawing groups are often incorporated to deepen the HOMO while retaining the mobility benefits of the Se atom.
The comparative analysis of BDT and BDS derivatives reveals a clear dichotomy driven by the heteroatom effect. BDT offers a robust, chemically stable, and versatile scaffold with deep HOMO levels suitable for high-voltage applications. BDS, through the incorporation of selenium, unlocks superior charge transport properties, lower reorganization energies, and extended spectral response, albeit with a trade-off in oxidative stability and synthetic cost.
The mechanism of electropolymerization for these fused systems is characterized by a rapid radical cation coupling sequence, pre-organized by strong π-stacking interactions unique to their planar geometry. The identification of π-dimer intermediates highlights the supramolecular nature of the polymerization process, suggesting that controlling the aggregation state of the monomer in solution could offer a handle to tune the morphology of the resulting polymer film.
Future research will likely converge on hybrid strategies. For instance, the use of BDT cores with selenium-containing side chains, or random copolymers of BDT and BDS, offers a pathway to synergistic materials that combine the high voltage/stability of sulfur with the high mobility/current of selenium. Furthermore, the development of new electrolyte systems, such as ionic liquids or boron-based Lewis acids, will continue to refine the electropolymerization process, enabling the fabrication of highly ordered, defect-free films for the next generation of bio-electronics and renewable energy devices [151].
4. Applications
BDT, BDF and BDS derivatives have found widespread applications in the field of optoelectronics, especially as organic semiconductors materials for devices such as organic light-emitting diodes (OLEDs) [152], organic field-effect transistors (OFETs) [153], organic photovoltaic cells [154], and nonlinear optical materials [155]. Numerous studies have been published in recent years highlighting their relevance in these areas. Interestingly, BDT derivatives have also been explored in supramolecular chemistry and as chemosensors for the selective detection of toxic heavy metal ions. Moreover, although less extensively investigated compared to optoelectronics applications, BDT and BDF systems have shown potential in the biological domain.
4.1. Organic Light-Emitting Diodes (OLEDs)
π-Conjugated polycyclic heteroaromatic compounds have been extensively studied in OLED devices, owing to their rigid π skeletons that generally confer effective luminescent properties, high carrier mobility and thermal stability. The first studies on the electroluminescence properties of BDT-1-based π-conjugated molecules 170–172 (Figure 3) were performed by K. Tanaka and co-workers starting in early 2000 [156,157].
The presence of a vinylene bridge between the two BDT-1 cores was fundamental for the emission properties of compounds 172, which were found to be highly fluorescent in solution and in thin film [156]. Different substituents on the bridging double C-C bond in 172 remarkably affected their luminescent features [156]. The flat system 172b, in which the resonance between the two BDT-1 cores was not perturbed, displayed quantum efficiency of fluorescence in solution (0.32) higher than those found for the twisted molecules 172c,d (0.05–0.045), in which the geometrical hindrance, arising from the presence of methyl or phenyl substituents, disturbed the resonance and reduced the transition probability. A preliminary single-layered device made of ITO/172b/Al:Mg was prepared, and a work function value of 5.62 eV was found for 172b by photoemission measurements [156]. More recently, OLED triplet emitters 173 and 174 (Figure 4) were reported as promising alternatives to metal–organic emitters, in which the “heavy-atom effect” hampered dissipation of electrically energy as heat [158].
Moreover, the effect of the conjugation on radiative and nonradiative relaxation of the triplet state in the two isomers 174 and 175 (Figure 4) was investigated by electroluminescence, quantum chemistry, and electron paramagnetic resonance spectroscopy [159]. The different position of the two thienyl rings significantly altered the effective conjugation path and induced different localizations of the spin density either on the phenazine unit or on the thienyl rings, while the phosphorescence was ensured by the contribution of the phenazine np* excited state. Although the thienyl rings were not necessary for generating phosphorescence, their presence increased the conjugation and then induced a red shift. The latter allowed for the development of emitters that can be used as dopants in organic semiconductor matrices.
In 2023, red thermally activated delayed fluorescence (TADF) emitters based on decorated phenazines 176, 177 and 178, 179 (Figure 5), bearing a donor-acceptor-donor (D-A-D) and a donor-acceptor-acceptor (D-A-A) structure, respectively, were synthesized and their photoluminescence (PL) and electroluminescence (EL) properties were investigated [160].
The D-A-A emitters 178 and 179 displayed better PL and EL properties than those of D-A-D ones, especially in terms of the relatively higher PL quantum yields and lower nonradiative rates. An effective intra- and intermolecular charge transfer was found in 178 and 179, due to intra- and intermolecular hydrogen bonds between the triphenylamine (D) group and the diphenylphosphine oxide (A) moiety. Moreover, the steric hindrance of the diphenylphosphine oxide efficiently inhibited concentration quenching. The device based on compound 179 exhibited the best performance (luminance max = 19,360 cd m^−2^, external quantum efficiency = 11.4% at 632 nm).
Benzodithienyl silane 180 (Figure 6), bearing a non-conjugated 3D geometry in which a tetrahedral silicon atom links two BDT-1 units through a dimethylsilyl bridge, was investigated as a promising semiconductive host material in devices [161].
Compound 180 crystallized into two monoclinic structures with aggregation-induced emission-like deep blue emission (390–397 nm) had a quantum yield up to 13%. Moreover, it was used as host material in green and blue emissive OLEDs to sensitize the green phosphor tris[2-phenylpyridine]iridium(III) (Ir(ppy)3) and the sky-blue bis2-(4,6-difluorophenyl)pyridyl-C2,Niridium(III) (FIrpic), respectively. While an optimal sensitization of the green Ir(ppy)3 emitter was achieved, the blue FIrpic emitter was instead only partially sensitized on account of the triplets being too close. On the other hand, the BDF-2 framework was used to develop a host material in blue phosphorescent OLED (PHOLED) devices [129]. Compound 136 (Figure 6) displayed high carrier mobilities (10^−3^ cm^2^V^−1^s^−1^) for both holes and electrons in the amorphous state, and, thanks to its high excited triplet-state energy level (ET = 2.77 eV), it was able to sensitize FIrpic (ET = 2.65 eV), thus demonstrating the suitability of 136 to build full-color PHOLED devices.
4.2. Organic Field-Effect Transistors (OFETs)
Thiophene-containing systems represent one of the most popular components used in OFETs, and numerous conjugated small organic molecules and polymers containing BDT-1 and BDT-2 frameworks have been synthesized and tested as semiconductors in OFETs.
4.2.1. Small Organic Molecules
Several studies on the use of air-stable BDT-1-based π-conjugated molecule 172b (Figure 3) as p-type semiconductors in OFETs were performed by K. Tanaka and co-workers [162,163,164,165,166]. The flat molecular plane of 172b significantly enhances its carrier mobility due to the strong interactions between adjacent molecules. Despite this potential, these studies clearly demonstrated that the maximum hole mobility is strongly influenced by the morphology and crystallinity of the vacuum-evaporated films [167].
The BDT-2-based π-conjugated molecule 55 (Figure 7) also showed promising electrical performances in organic thin film transistors [90]. In particular, though both the BDT-2 core and the extended π-conjugated styryl systems were not planar, the molecular arrangement in 55-based thin films promoted an efficient charge transport across the silicon oxide semiconductor interfaces in an organic thin film transistor configuration. The dithienonaphthalene 102b (Figure 7) also worked as p-type semiconductor in OFET [116], with better performance than those of thiophene- and thienothiophene-based derivatives 181 and 182 (Figure 7) [168].
Alternatively, the push–pull semiconductor system 183 (Figure 7), containing the BDT-2 scaffold as donor alternated to the benzothiodiazole core as acceptor, displayed hole mobility (1.4 × 10^−2^ cm^2^ V^−1^ s^−1^) in OFET devices [169].
Numerous π-extended polycyclic aromatic hydrocarbon systems incorporating the BDT-1 core were found to be stable and promising low-molecular weight organic semiconductors in OFETs [41,52,68,71,72,73]. OFET devices of alkylated phenazines 184a, 185a and 186 incorporating BDT-1 and/or BDT-2 cores were fabricated by vacuum deposition, and their performances were compared with those of anthracene analogs 184b and 185b (Figure 8) [170].
Thin films of 184a, 185a and 186 showed nearly the same crystallinity and then similar FET features (around 10^−6^ cm^2^ V^−1^ s^−1^ when deposited at 75 °C), regardless of the position of sulfur atoms. On the other hand, lower FET mobilities were obtained for BDT systems than those of anthracene analogs 184b and 185b (around 10^−2^ cm^2^ V^−1^ s^−1^) [58,68], presumably due to lower donor abilities and the larger reorganization energies of the nitrogen-containing systems. BDT-1 and BDT-2-based semiconductors with ambipolar charge transport properties were also synthesized, and their electrical behavior was examined [60,74,78]. The ambipolar charge transport in field-effect transistor devices was observed for the fused heteroaromatic compounds incorporating donor/acceptor structures, including molecule 187 [78], 188 [74] and fused dithieneonaphthothiadiazoles 189 and 190 [60] (Figure 9).
4.2.2. Polymers
Besides small organic molecules, π-conjugated semiconducting polymers containing the BDT-1 and BDT-2 framework were found to be promising high-performance OFETs. The introduction of the BDT-2 core into a semiconducting polymer backbone 191 (Figure 10) led to an active material in OFET [171]. This polymer displayed high charge-carrier mobility (0.5 cm^2^ V^−1^ s^−1^) and was found to be suitable for application on flexible substrates (i.e., PET film). The curvature of the BDT-2 core ensured the best compromise between solubility and aggregation tendency towards the quick formation of highly ordered films.
An alternative approach towards semiconducting polymers for use in high-performance OFETs relies on the copolymerization of donor and acceptor units to yield D/A copolymers with high charge-carrier mobility. BDT frameworks were found in π-extended systems which were employed as building blocks to create alternating D/A copolymers such as 192 [172] and 193 [173] (Figure 10). In these systems, the strong D-A interactions in combination with the highly coplanar polymer backbone guaranteed π–π stacking self-assembly and a compact solid-state packing associated with high charge-carrier mobility in OFETs, while the presence of long branched alkyl chains (e.g., 2-octyldodecyl) improved the solubility in organic solvents.
π-Conjugated D/A polymers containing BDT and BDS cores incorporated into one or two-electron-poor imide units, such as 194 [139], 195 [174], 196 [174] and 197 [175] (Figure 11), were also tested as semiconductors in OFET devices.
The nature of chalcogen atoms in combination with the design of the copolymers 195 and 196 significantly affected their charge transport properties. The average/maximum electron mobility of BDS-based polymer 195b (0.005/0.01 cm^2^ V^−1^ s^−1^) were an order of magnitude higher than those of BDT-based polymer 195a (4.2 × 10^−4^/7.1 × 10^−4^ cm^2^ V^−1^ s^−1^). Conversely, the average/maximum electron mobility of BDT-based copolymer 196a (0.003/0.005 cm^2^ V^−1^ s^−1^) was an order of magnitude higher than that of BDS-based polymer 196b (3.5 × 10^−4^/5 × 10^−4^ cm^2^ V^−1^ s^−1^). Overall, the electron mobility of most of these systems is comparable to the classic n-type semiconductor (i.e., PC_71_MB ca. 10^−3^ cm^2^ V^−1^ s^−1^) [176]. The polymer 198 (Figure 11), containing a strong electron-deficient dithiene-fused quinoxalineimide, displayed unipolar n-type transport character with an electron mobility of 0.25 cm^2^ V^−1^ s^−1^ in OFETs [177].
4.3. Organic Solar Cells (OSCs)
Functionalized BDT systems along with BDT- and BDS-based polymers have been found to be promising organic semiconductors for the development of bulk heterojunction (BHJ) OSCs, dye-sensitized solar cells (DSSCs), and perovskite solar cells (PSCs).
4.3.1. Bulk Heterojunction (BHJ) OSCs
BHJ solar cells, reported for the first time by Yu and co-workers in 1995 [178], are still one of the most promising OSCs in photovoltaic technology [179]. They are made of an active layer formed by a conjugated organic small molecule or a polymer as a donor (p-type semiconductors) and a fullerene derivative or a non-fullerene small molecule or polymer as an acceptor (n-type semiconductor). BDT-1 and especially BDT-2 frameworks were used in the form of small molecules or polymer species, either as p-type or n-type semiconductors in BHJ OSCs. More recently, a few examples of BDS-2-based polymers as donors in BHJ OSCs were also reported.
Polymer Donors Based on BDT and BDS
The donor–acceptor (D-A) copolymer D18 (Figure 12), which alternates the electron-donating (D) benzo[1,2-b:4,5-b′]dithiophene and the electron-accepting (A) BDT-2 fused-ring benzothiadiazole, represents one of the most interesting high-performance donor polymers in BHJ systems [180]. D18 was proposed for the first time by Ding and co-workers [181] and, when it was blended with the small acceptor molecule Y6 (Figure 12), a remarkable power conversion efficiency (PCE) up to 18.22% was achieved. More recently, thanks to their high device performances and suitable morphological characteristics, polymer donors based on D18 and its analogs were also successfully applied in all-polymer solar cells, obtaining PCE up to 19% [182,183,184].
Fullerene-based BHJ solar cells were also fabricated using donor copolymers containing the BDT-1 framework, such as the A-D structures 199 (Figure 12) [76], made of BDT-1 containing imide (A) and 2,2′-bithiophene units (D), and the structure D-A_1_-D-A_2_ 200 (Figure 12), composed by the benzothiadiazole acceptor (A_1_), the BDT-1 fused-system (A_2_) and the thiophene ring as donor (D) [75]. However, in both cases, modest PCEs were obtained (2.45–6.21%).
In 2022, a comparative study demonstrated that the orientation of the two thiophene rings in the acceptor unit of donor copolymers 201 and 202 (Figure 13) significantly affected their spectral and morphological properties as well as their efficiency in BHJ devices using Y6 as the acceptor [102]. Indeed, while the device fabricated with the BDT-2-based copolymer 201 provided a PCE of 15.05%, the use of the BDT-1-based copolymer 202 afforded devices with almost no solar cell performance.
On the other hand, wide-bandgap D-A donor copolymers 203a,b (Figure 13) were employed to develop BHJ solar cells blended with Y6 as a non-fullerene acceptor [185]. In this case, the BDT-2 scaffold represented the donor unit (D) while the BDT-1 portion was the acceptor one (A), and a PCE up to 16.19% was achieved in the ternary devices made of a mixture of 203a:203b blended with Y6.
Examples of BDT-1 [186], BDT-2 [104,187,188] and BDS-2 [140,189] cores used as donor moiety in the donor-acceptor (D-A) copolymers were also reported as donor polymers in BHJ systems, though low to moderate PCE values were achieved (up to 11%).
Acceptors Based on BDT
As far as the use of BDT-based systems as n-type semiconductors in BHJ solar cells, non-fullerene small-molecule-acceptor 204 [190] and the acceptor polymer species 205 [191], (Figure 14), both incorporating the BDT-2 on the quinoxaline-fused core were used to develop OSCs with PCE ranging from 14.14 to 17.05%.
Interestingly, when the two isomeric BDT-2 and BDT-1 unit were fused at the bay position of perylenediimide skeleton, the corresponding n-type organic semiconductors 206a,b (Figure 14) displayed different device performances in non-fullerene OSCs, and in this case the higher PCE value was also achieved by the BDT-2-based acceptor 206a (4.44%) in comparison with that provided by the BDT-1 system 206b (2.98%) [192].
4.3.2. Dye-Sensitized Solar Cells (DSSCs)
After the study reported by Gratzel and O′regan in 1991 [193], DSSCs gained a lot of attention as a low-cost and renewable energy source in photovoltaic technology [194]. The dye sensitizer represents the key element of a DSSC, and it is responsible for the light-harvesting and charge-separation processes. Organic dye is generally composed of an electron donor unit (e.g., alkoxyl substituted triarylamines) and an electron acceptor unit (e.g., cyanoacrylic acid), which are covalently connected through a π-conjugated spacer, whose structure significantly affects the photophysical properties of the dye. In 2012 [195] and 2013 [196], the BDT-1 framework was used as a π-spacer in dye sensitizers 207 (Figure 15), and power conversion efficiencies (η) ranging from 3.6 to 5.6% in a liquid cell were achieved. These similar values indicate that the nature of the heteroaromatic linkers (i.e., benzene, thiophene and thiazole) does not have an appreciable influence on efficiency. In 2017, the BDT-2 core was employed as a π-spacer to yield sensitizers 208 (Figure 15), in which a heteroaromatic fragment between BDT-2 and the acceptor unit was inserted to improve the light harvesting of the dyes. PCE values higher than 9% were obtained using the I^−^/I^3−^ or Co(phen)3^2+/3+^ as an electrolyte in the presence of chenodeoxycholic acid as co-adsorbent [197].
The near-infrared co-sensitizer 209 (Figure 15), containing the electron-withdrawing BDT-1 framework, and the co-sensitizer 210 (Figure 15) were mixed to set quasi-solid-state DSSCs, which exhibited PCE up to 8.04% and long-term stability (1000 h under continuous light soaking) [198].
More recently, Zhong and co-workers developed a family of metal complexes containing the sulfur coordination of the BDT-2 core, which were used as monomers to create dye sensitizer copolymers for DSSCs [199,200,201]. These metal-based polymeric sensitizers 211–213 (Figure 16) are characterized by a D-A1-π-A2 motif, in which benzo[1,2-b:4,5-b′]dithiophene cores were used as electron donors (D), the metal complexes containing the BDT-2 ligand acted as auxiliary electron acceptors (A1), and 8-quinolinol derivatives were used as π-bridge and electron acceptor (A2) units.
Five metals (i.e., Ni, Cu, Zn, Cd and Hg) were selected to synthesize the corresponding polymers and to study their performance in DSSC devices. As a general trend, PCEs increased with the increasing radii of Ni(II), Cu(II), Zn(II), Cd(II) and Hg(II), and the best photovoltaic performance was achieved with Hg(II)-based polymers (10.96 [199], 11.78 [200] and 12.89% [201]).
Although the metal ions displayed the same charge, their increasing ionic radius strengthened the coordination with the sulfur of BDT-2. This, in turn, enhanced both the electron-withdrawing capability and the intramolecular electron transfer of the metal-based acceptors A1. This stronger interaction improved the electron absorption capacity and the push–pull electron balance, narrowed the polymer bandgap, induced the redshift of λ_max_, and boosted photovoltaic performance.
4.3.3. Perovskite Solar Cells (PSCs)
Perovskite solar cells (PSCs) represent younger devices than BHJ OCs and DSSCs, since the first example of PSCs was reported in 2009 by Miyasaka and co-workers, in which a PCE of 3.8% was obtained [202]. Since this work, PSCs have rapidly attracted much attention due to their impressive capability of converting solar energy into electricity with skyrocketing PCEs, currently exceeding 25%. Generally, PSCs consist of five parts, including a transparent conductive metal electrode, a hole-transporting material (HTM), a perovskite light absorber layer, an electron transport layer, and a black electrode of carbon or noble metal. The HTM collects and transports photo-generated holes between the absorption layer and electrode of PSCs, realizing effective separation of electrons and holes, which is an essential point to enhance PCE of PSCs [203]. Organic HTMs based on conductive small molecules or polymers have been widely investigated in high-performance PSCs, and the spiro-OMeTAD (Figure 17) still represents one of the most advantageous HTMs in PSCS technology, though some obstacles towards large-scale applications have led to further exploration of alternative and efficient HTMs.
Small-molecule semiconductors containing the BDT-1 and BDT-2 frameworks were employed as HTMs in PSCs, demonstrating great potential thanks to their high hole mobility, thermal stability, and tunable molecular structures. Naphto BDT-1 [64,204] and BDT-2 [116] were incorporated as π-bridges to connect the two electron-rich triphenylamine (TPA) units in HTMs 214 (Figure 17) and 102b (Figure 7), respectively. For compound 214, the PCE reached considerable values up to 18.8%, slightly higher than the spiro-OMeTAD (18.1%). More recently, HTM 215 (Figure 17), bearing a carbon-carbon double bond as π-spacer between the BDT-2 core and the triarylamine units, showed very good PCEs up to 17.2% [205], without any device oxidation treatments, which are generally required for spiro-OMeTAD.
Alternatively, π-bridges made of BDT-1 [206] and BDT-2 [207] incorporated into a quinoxaline core provided D-A-D-type HTMs 216 and 217 (Figure 17), respectively, with the TPA as donors (D) and the quinoxaline derivatives as acceptor units. These HTMs displayed photovoltaic performances competitive with spiro-OMeTAD, and PCEs over 20% with excellent long-term stability were achieved by PSC devices using the dopant-free 216 (21.03%) and the doped HTM 217 (20.52%).
Finally, three D-A-D dopant-free HTMS 218 (Figure 17) were designed and tested in PSCs, in which two TPA donor units were linked to different acceptors made of fused BDT-2 chalcogenadiazoles (i.e., furazane, thiadiazole and selenadiazole) [208]. The nature of the chalcogen atom in the diazole framework had a remarkable effect on the optical, electrochemical, and charge-transport features of 218, and the best photovoltaic performance was obtained in dopant-free PSCs made of selenium-containing HTM 218c (PCE for 218c of 15.09% vs. 13.31% for oxygen derivative 218a and 11.65% for sulfur derivative 218b).
4.4. Nonlinear Optical (NLO) Materials
Nonlinear optical (NLO) compounds generally include donor and acceptor groups linked through a π-conjugated spacer, and, among them, organometallic species exhibit interesting NLO properties thanks to the coordination of the metal atom to conjugated ligands that promote effective charge transfer transitions [209]. The BDT-1 framework was used as a ligand for coordination to the metal center of [Fe_2_(η^5^-C_5_H_5_)2(CO)2(μ-CO)(μ-C–CH_3_)]^+^[BF_4_]^−^ [210] and a family of cyclopentadienyl iron/ruthenium derivatives [211].
First studies on the quadratic hyperpolarizabilities (β) of iron and ruthenium complexes 219 (Figure 18) were performed by hyper-Rayleigh scattering measurements at 1500 nm, obtaining a value of β0 = 19 × 10^−30^ esu for 219a, and irrelevant values for 219b,c [211].
Besides organometallic complexes, the development of organic molecules with donor–acceptor architecture and a π-conjugated bridge also represents an effective approach towards second-and/or third-order NLO materials with enhanced optical hyperpolarizabilities [212]. Two intramolecular charge-transfer (ICT) compounds 117a,b (see Scheme 36), in which the cyano groups acted as acceptors while the angular-shaped structure of the π-conjugated BDF-2-based bridge provided efficient ICT processes, exhibited low-dimensional microstructures with significantly distinctive linear and nonlinear optical properties, as a consequence of diverse orientations of their transition dipoles and self-assembled structures [122]. More recently, the intramolecular boron-locking strategy was employed in a series of BDT-2-based donor-acceptor compounds, such as 220 and 221 (Figure 18), to evaluate how torsion angles θ_1_ and θ_2_ between the donor unit (i.e., the triphenylamine) and the boron-locking acceptor affected the first hyperpolarizability (β) value [213]. In particular, the decrease in the torsion angles in 221 significantly increased the β value by up to 94%, likely due to the lower excited energy of the key excited state and enhanced charge transfer from the triphenylamine group to the BDT-pyridine moiety.
4.5. Self-Assembly
Shape-persistent macrocycles (SPMs) are promising compounds in the field of supramolecular chemistry and self-assembly because they present an inner cavity in the nanometer regime, and the building blocks of the ring are rather rigid and connected in such a way that the whole structure cannot collapse. Due to their rigidity, SPMs can be functionalized independently in their interior and exterior, and the orientation of the side groups remarkably influences the properties and applications of SPMs [214]. SPMs containing thiophene-based units are interesting systems to build self-assembled monolayers at the solid/liquid interface and for application in photovoltaics. The most significant contribution in this field was provided by S. Höger and co-workers, who developed different families of π-expanded shape-persistent macrocycles containing BDT-1 [215,216,217] and BDT-2 [218,219,220] units.
Macrocycles 222 and 223 (Figure 19), containing phenylene-ethynylene-butadiynylene backbone alternated to naphtho[2,1-b:3,4-b′]dithiophene [218] and benzo[1,2-b:4,3-b′]dithiophene [216] units, respectively, yielded 2D ordered arrays on highly oriented pyrolytic graphite (HOPG) by self-assembly under ambient conditions. The macrocycle arrays ordered from 222 represented a suitable template for the epitaxial co-adsorption of metallacycles, thanks to the presence of electron rich BDT-2 units that favored the electronic interactions with the electron withdrawing guest molecules, while macrocycles 223 afforded supramolecular empty helical nanotubes in the liquid crystalline mesophases.
Höger’s group also reported the synthesis of SPMs [224]n (n = 3–6) (Figure 19) based on BDT-1 corner pieces connected through phenylene-ethynylene-butadiynylene units, in which the ring sizes and the extra-annular alkoxyl chains ensured enough solubility and elastic deformability of the systems [215]. All macrocyclic oligomers, when adsorbed under ambient conditions at the interface of diluted solutions of [224]n in 1,2,4-trichlorobenzene (TCB) as solvent (10^−4^–10^−7^ M) and HOPG as substrate, assemble to highly ordered 2D patterns, supporting the formation of molecular porous and dense long-range ordered patterns of complementary shapes. Thereafter, A. Bedi and S. Zedi synthesized a macrocycle 225 (Figure 20), containing two BDT-1 units linked through thienyl ethylene spacers [217]. This macrocycle underwent self-assembly in the solid state, forming microfibers of the length of several μm and thickness of ∼400 nm on the Si/SiO_2_ surface. The SPM donor-acceptor hybrid system 226 (Figure 20) was also synthesized [219], in which the electron rich macrocycle backbone, made of four naphto[1,2-b:4,3-b′]dithiophene units connected through two phenylene rings, was decorated with two extra-annular electron poor perylene bisimide groups. This system formed supramolecular 1D aggregates at the TCB/HOPG interface, mainly governed by the strong attractive forces between the acceptor perylene bisimide moieties. Moreover, a deep study of its charge separation properties through different electron paramagnetic resonance (EPR) techniques in combination with light excitation showed the presence of intermolecular and intramolecular charge separation, and the observed radical pair states induced upon illumination made hybrid systems like 226 as promising semiconductors in organic photovoltaic cells [220].
More recently, BDT-1 based tetrathienoanthracenes 227 (Figure 20) were synthesized and used as planar discotic fragments for the development of π-conjugated discotic liquid crystal (DLC) systems as hole-transporting materials bearing a supramolecular columnar architecture [221]. The great tendency of 227 to align homeotropically in the columnar mesophase over a large area in space-charge limited current (SCLC) cells in combination with the strong co-facial π–π interactions and multiple S^…^S contacts between the tetrathienoanthracene core ensured a remarkably high hole mobility (4.22 cm^2^V^−1^s^−1^) for this DLC system in SCLC device.
4.6. Chemosensors
Methods based on fluorescent and colorimetric chemo-sensory materials for the selective recognition of biologically and environmentally toxic heavy metal ions (e.g., Hg^2+^, Pb^2+^) has been receiving much attention due to their excellent sensitivity and selectivity, along with their response time, local observation, nondestructive character, economic nature, and synthetic simplicity.
In particular, the development of highly sensitive chemosensors with fluorescence turn-off or turn-on sensing mechanism for the specific recognition of toxic metal ions analyte represents an important objective in chemistry, biology and medicine. BDT-1-based small molecular organic and π-conjugated polymeric chemosensors 228 and 229 (Figure 21), respectively, with neighboring nitrogen and sulfur heteroatoms as chelating sites for the selective detection of bivalent cations were reported [222,223]. Organic molecules 228 displayed remarkable sensitivity towards Pb^2+^ over the other metal ions in aqueous solutions [222], while π-conjugated polymer 229 was found to be a suitable system for both colorimetric and ratiometric detection of Hg^2+^ and the fluorometric detection of Zn^2+^ through a fluorescence turn-on response with an enhanced fluorescence lifetime in the presence of Zn^2+^ [223]. Moreover, the 229-Zn complex allowed the selective colorimetric detection of I^−^ over other anions.
Similar polymers 230 (Figure 21) acted as fluorescence sensors for the selective sensing of biological and environmentally relevant Cu^2+^ [224], since the presence of both hard N and soft S donors made 230 efficient fluorescence sensors towards slightly acid transition metal cations such as Cu^2+^. Polymer 230b exhibited outstanding sensitivity and selectivity towards Cu^2+^ by emission quenching via photoinduced electron transfer, and its polymeric film was also investigated as a thin-film polymeric sensor for application in on-site detection of Cu^2+^. In this case, the sensing ability of these polymers was strictly dependent on the electronic nature of the substituents on the phenyl ring of 230, since 230b, with a p-bromophenyl pendant, succeeded over the toluene-based congener 230a towards Cu^2+^ sensing. Emissive and well-defined π-conjugated copolymers 231 (Figure 21) were found to be low-cost and highly selective and sensitive turn-off fluorescent probes for the selective detection of Hg^2+^, with a limit of detection up to ppb level (40–50 ppb) in semi-aqueous environment [225]. The study of optical probes for the qualitative or quantitative detection of anions also represents an important target for health and environment. The anion-responsive BDT-2-based molecule 232 (Figure 21) exhibited especially strong interaction with carboxylate, dihydrogen phosphate and cyanide anions, through H-bonding with the two N−H bonds of sulfonamide groups [226].
4.7. Biology
Although BDT and BDF frameworks have been extensively studied for their optoelectronic properties in materials science, their application in biological domain, while less common, has also been documented.
Some BDF-1 and BDF-2 derivatives displayed cytotoxic activity against different human cell lines. The in vitro cytotoxicity of BDF-1 systems 233 (Figure 22) against human hepatocellular liver carcinoma cell line (HepG2) and their antioxidant activity using 2,2-diphenyl-1-picrylhydrazyl (DPPH) and 2,2′-azino-bis-3-ethylbenzthiazoline-6-sulphonic acid (ABTS) bioassays were evaluated and compared with those of three naturally occurring naphthoquinones (i.e., juglone, lawsone, and plumbagin) [227].
Compounds 233b,c showed similar and higher scavenging activity in DPPH assay in comparison with that of natural naphthoquinones, while comparable activity for all molecules was observed in ABTS bioassay. Conversely, no cytotoxic activity was observed against HepG2 cell line for derivatives 233b,c at all concentrations (up to 100 mg/mL), but a significant anti-proliferative activity was revealed for BDF-1 233a (IC_50_ 26.05 mg/mL), higher than juglone (IC_50_ 48.61 mg/mL) and plumbagin (IC_50_ 58.63 mg/mL).
A set of BDF-1 and BDF-3 derivatives were tested in vitro for the inhibition of human breast cancer cell lines T-47D and MCF-7 [228]. Compounds 234 and 235 (Figure 22) were found to be the most promising selective estrogen receptor modulators (SERMs) with IC_50_ values similar to that of the tamoxifen.
BDT derivatives were also designed and applied in biological field, demonstrating potential both as anticancer agents and as fluorophores for bioimaging applications. The antiproliferative activity of BDT-1 derivative 236 (Figure 23) in cultured rat aortic smooth muscle cells (SMCs) and in the human NCI-H460 lung tumor cell line was comparable to that achieved by Amonafide (Figure 23) that was included as a reference compound [229]. DNA intercalating assay demonstrated that 236 did not directly interact with DNA, so its antiproliferative effect was not a consequence of its DNA intercalating activity.
The inhibitory activities of various BDT-2 systems against alpha-glucosidase, urease, and the free radical production were evaluated [230]. Among them, compound 237 (Figure 23) displayed significant DPPH radical scavenging activity and exhibited promising alpha-glucosidase inhibition relative to other tested molecules. Structure-activity relationship studies suggested that the presence of the two chlorine atoms play a key role in its activity.
The incorporation of a BDT-2 and a BDT-1 framework onto the structures 238 and 239 (Figure 23), respectively, enabled the development of novel fluorophores emitting in the second near-infrared (NIR-II) window for biomedical applications [231]. Notably, 239-based nanoparticles exhibited bright NIR-II emission, allowing in vivo imaging with high signal-to-background ratios and facilitating long-term stem cell tracking for acute lung injury detection. Compound 240 also displayed an absolute QY of 0.4% with extension to 1400 nm, making it highly suitable for NIR-II bioimaging [232]. Furthermore, cerebrovascular function, including cerebral blood flow and vascular reactivity under various conditions, was accurately quantified.
BDT-1 was also employed as a ligand in cationic Ru(II) complex 241 (Figure 24), which showed higher cytotoxicity than cisplatin against human leukemia cancer cells (HL-60 cells) [233]. This activity could be ascribed to some DNA interactions as evidenced by CD, electrophoretic mobility and AFM studies. The most plausible interactions rely on weak ligand–DNA interactions through hydrogen bonds.
More recently, incorporation of a BDT-1 framework into a classic DNA–intercalative Ru(II) complex with chemotherapeutic activity yielded a two-photon absorption Ru(II) complex 242 (Figure 24) [234]. This complex is localized to both mitochondria and nuclei, enabling the regulation of DNA-related chemotherapy mechanisms such as DNA topoisomerase and RNA polymerase inhibition. It demonstrated remarkable phototherapeutic efficacy with minimal toxicity to normal liver and kidney cells in vitro, and in vivo it showed high antitumor activity against malignant melanoma and cisplatin-resistant NSCLC, achieving 100% mouse survival, low toxicity to normal cells, and minimal residual tumor rates.
4.8. Miscellaneous
4.8.1. BDT-Based Porous Organic Polymers (PSCs)
Porous organic polymers (POPs), especially conjugated microporous or porous polymers (CMPs or CPPs), have received increasing attention due to their remarkable and designable properties, such as chemical and thermal stability, tunable band gap, large surface area and adjustable porosity due to their porous nature. All these features make them highly attractive systems for applications in different fields, including gas storage and separation, energy storage, water purification, photoelectricity, chemosensors, organic electronics and heterogenous catalysis (e.g., photocatalysis) [235]. A wide range of π-conjugated organic molecules have been used to build CMPs and CCPs, including nitrogen-, oxygen- and sulfur-containing heteroaromatic moieties such as tetrathienoanthracenes (TTA), naphthodithiophenes and benzodithiophenes. In 2021, a 3D π-conjugated microporous polymer 243 (Figure 25), containing the BDT-1-based tetrathienoanthracene (TTA), was synthesized through a bottom-up approach, and used as a heterogeneous organocatalyst for visible light-promoted organic reactions [236]. The great photoredox features along with its excellent stability and mesoporous structure made the polymer 243 a highly efficient (80–98% yield for 29 examples) and reusable (10–12 times) heterogenous photocatalytic system for model organic transformations, including the oxidative C–C bond formation between N-aryltetrahydroisoquinolines and nitroalkanes or ketones, and the coupling of phenylenediamines with aldehydes towards the synthesis of benzimidazoles.
In 2023, the TTA framework was also employed as a sulfur-rich and π-conjugated skeleton to design and synthesize star-type 3D CPPs that represented promising candidates for photocatalytic CO_2_ conversion [237].
The polymer arising from the Sonogashira–Hagihara coupling (SHC) between Br-TTA (Figure 25) with the more flexible ethynylphenyl methane structure (EtPhM, Figure 25) exhibited a larger specific surface area, a better CO_2_ adsorption capacity, and a lower energy barrier for the rate-determining step of CO_2_ photoconversion than those observed for the more rigid pyrene-based polymer formed by SHC between Br-TTA and EtPy (Figure 25). Indeed, the EtPhM-based polymer provided a higher CO evolution rate and selectivity (up to 322.05 μmol g^−1^ h^−1^ and 99.26%, respectively).
4.8.2. BDT-Based Multifunctional Materials
BDT-1 derivatives 244 (Figure 25), differing in the position of the nitrogen atom in the pyridine rings, were identified as multifunctional materials with peculiar self-assembly behavior [238]. In particular, compound 244a, with nitrogen in the para position, self-assembled into a columnar liquid-crystalline phase and exhibited significant, red-shifted absorption and emission peaks due to enhanced conjugation. Further, 244a also formed gels in different organic solvents, and when doped with L-tartaric acid, gave a gel with intensified yellow emission, suitable for white light-emitting diodes (WLEDs) production. Moreover, the protonation of pyridine in 244 enabled acid–base vapor detection, and both compounds exhibited pronounced acidochromic behavior, making them promising indicators for acid–base sensing in the film states.
5. Conclusions and Perspectives
This review underscores, for the first time, the significance of isomeric benzodithiophene (BDT), benzodifuran (BDF) and benzodiselenophene (BDS) analogs of phenantrene as key members of chalcogen-containing tricyclic β-fused systems. Their distinctive angular architecture imparts properties markedly different from those of their anthracene-like counterparts, while the spatial arrangement of heteroatoms within these frameworks emerges as a critical determinant of structure–property relationships. These features influence not only the behavior of individual molecules but also their performance within polymeric architectures, where the nature of the heteroatom plays a pivotal role in modulating electronic characteristics.
BDT derivatives emerge as the most prominent class discussed in this review, owing to the breadth and versatility of synthetic methodologies developed for their construction. Robust and modular strategies for assembling BDT frameworks have enabled extensive structural diversification, facilitating downstream functionalization and integration into advanced material architectures. Moreover, BDT scaffolds are typically obtained as stable frameworks amenable to post-synthetic modification, underscoring their higher stability and synthetic flexibility than BDF derivatives. Indeed, most reported protocols for synthesizing BDF derivatives typically afford a single, well-defined compound and frequently yield pre-functionalized cores, thereby limiting opportunities for structural diversification. On the other hand, furan has recently emerged as a privileged building block among group 16 heterocycles, driven by its biodegradability and renewable sourcing [239]. Thus, the development of innovative synthetic strategies for furan-fused π-conjugated systems represents a promising approach toward sustainable materials design. The development of synthetic strategies for BDS derivatives is still at an early stage, with only a limited number of reported procedures that typically afford low yields. Nevertheless, efficient post-synthetic functionalization of the BDS core—such as functionalization of α-positions of selenium atoms and subsequent Pd-catalyzed coupling reactions—has been successfully demonstrated, highlighting significant opportunities for further development.
Among the applications reported for BDT, BDF, and BDS derivatives, optoelectronics stands out as the most extensively explored field, with numerous examples of significant relevance. The synthetic flexibility and stability of BDT frameworks have enabled a wide range of advanced device architectures, including OLEDs, OFETs, and photovoltaic systems. This versatility is further enhanced by the ease of oligomerization and polymerization of the BDT scaffold, which affords high-performance BDT-based polymers as semiconductors in bulk heterojunction organic solar cells. Controlled functionalization strategies allow for fine-tuning of electronic properties, reinforcing the pivotal role of BDT scaffolds in bridging molecular design with practical implementation. Looking ahead, the implementation of more sustainable synthetic procedures—aligned with the principles of green chemistry—will be essential to ensure that BDT derivatives become truly competitive and viable for large-scale applications.
Although less developed, BDT and BDF derivatives exhibit promising biological potential, functioning as cytotoxic agents and fluorophores for NIR-II bioimaging when appropriately engineered. This demonstrates that compounds traditionally confined to optoelectronic applications can gain attention for their potential roles in biological systems. Such emerging trends exemplify the growing interplay between materials science and life sciences, opening avenues for multifunctional platforms that integrate electronic performance with biomedical relevance.
On the other hand, BDS frameworks—owing to their high polarizability and tunable electronic characteristics—represent attractive candidates for future optoelectronic applications. Post-synthetic functionalization offers significant opportunities for tailoring electronic structures, paving the way for advanced materials in organic semiconductors, photovoltaic devices, and nonlinear optical systems. Expanding the synthetic toolbox to improve yields and structural diversity will be essential to fully exploit the potential of BDS derivatives in next-generation technologies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ye S. Lotocki V. Xu H. Seferos D.S. Group 16 conjugated polymers based on furan, thiophene, selenophene, and tellurophene Chem. Soc. Rev.2022516442647410.1039/D 2CS 00139 J 35843215 · doi ↗ · pubmed ↗
- 2Jeffries-El M. Kobilka B.M. Hale B.J. Optimizing the Performance of Conjugated Polymers in Organic Photovoltaic Cells by Traversing Group 16Macromolecules 2014477253727110.1021/ma 501236 v · doi ↗
- 3Katritzky A.R. Ramsden C.A. Joule J.A. Zhdankin V.V. Handbook of Heterocyclic Chemistry 3rd ed.Elsevier Amsterdam, The Netherlands 2010 Chapter 2.387138
- 4Ashraf R.S. Meager I. Nikolka M. Kirkus M. Planells M. Schroeder B.C. Holliday S. Hurhangee M. Nielsen C.B. Sirringhaus H. Chalcogenophene comonomer comparison in small band gap diketopyrrolopyrrole-based conjugated polymers for high-performing field-effect transistors and organic solar cells J. Am. Chem. Soc.20151371314132110.1021/ja 511984 q 25547347 · doi ↗ · pubmed ↗
- 5Tsuji H. Nakamura E. Design and Functions of Semiconducting Fused Polycyclic Furans for Optoelectronic Applications Acc. Chem. Res.20175039640610.1021/acs.accounts.6b 0059528165719 · doi ↗ · pubmed ↗
- 6Zander M. The Intra-annular Internal Heavy-atom Effect on the Fluorescence and Phosphorescence Properties of Oxygen, Sulphur or Selenium Containing Heterocyclic Systems Related to Dibenzo[b,n]perylene Z. Naturforschung A 1989441116111810.1515/zna-1989-1113 · doi ↗
- 7Zander M. Kirsch G. On the Phosphorescence of Benzologues of Furan, Thiophene, Selenophene, and Tellurophene. A Systematic Study of the Intra-annular Internal Heavy-atom Effect Z. Naturforschung A 19894420520910.1515/zna-1989-0306 · doi ↗
- 8Yadav V.K. Relative Aromaticity of Pyrrole, Furan, Thiophene and Selenophene, and Their Diels-Alder Stereoselectivity Steric and Stereoelectronic Effects in Organic Chemistry Springer Cham, Switzerland 202110.1007/978-3-030-75622-2_9 · doi ↗