Essential Role of LapD in the Absence of Cardiolipins
Satish Raina, Akshay Maniyeri, Aravind Ayyolath, Gracjana Klein

TL;DR
This study explores how the absence of cardiolipins in E. coli affects bacterial survival and reveals the regulatory role of LapD in lipid synthesis.
Contribution
The study identifies LapD as a key regulator linking LPS assembly and fatty acid/phospholipid synthesis in E. coli.
Findings
CL synthesis is conditionally essential in ΔlapD bacteria.
Loss-of-function mutations in cdsA and pgsA bypass lethality by reducing acidic phospholipids.
Overexpression of accC and glnB overcomes Δ(lapD clsA) lethality by inhibiting fatty acid/PL synthesis.
Abstract
To maintain the integrity of the outer membrane of Gram-negative bacteria, such as Escherichia coli, the levels of two essential components, phospholipids (PL) and lipopolysaccharide (LPS), are tightly regulated, although the underlying molecular mechanisms are unclear. E. coli synthesizes three main PLs, including essential phosphatidylethanolamine and phosphatidylglycerol and nonessential cardiolipin (CL). We showed that CL synthesis is conditionally essential in ΔlapD bacteria. Using this synthetic lethal phenotype, we isolated suppressors that rescued growth at elevated temperatures. We showed that loss-of-function mutations in cdsA encoding CDP-diglyceride synthetase, and pgsA, which encodes phosphatidylglycerophosphate synthase, bypass this lethality. Such mutations reduce the relative abundance of acidic phospholipids, which are otherwise elevated in Δ(lapD clsA) bacteria, and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
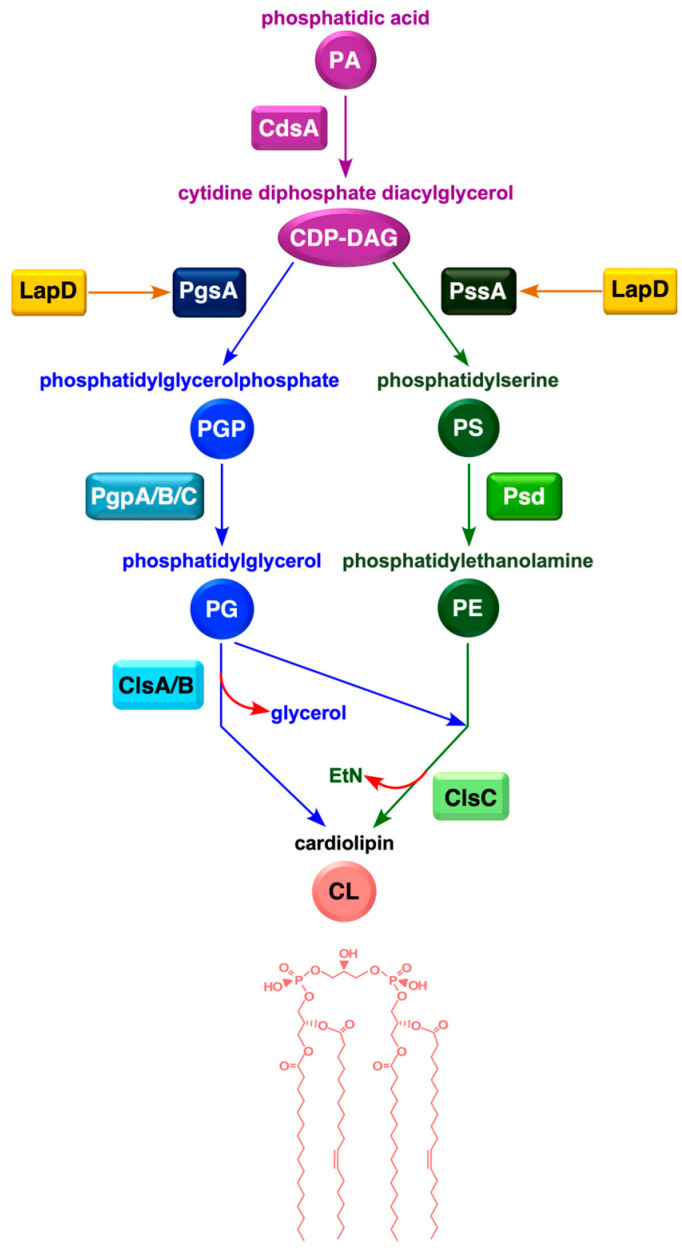 Figure 1
Figure 1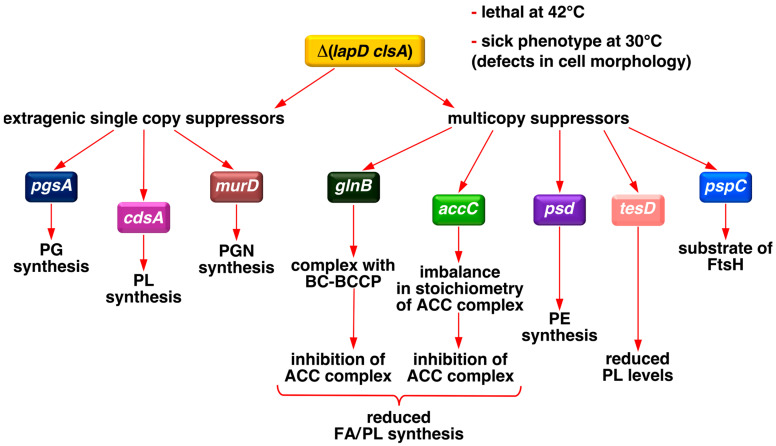 Figure 2
Figure 2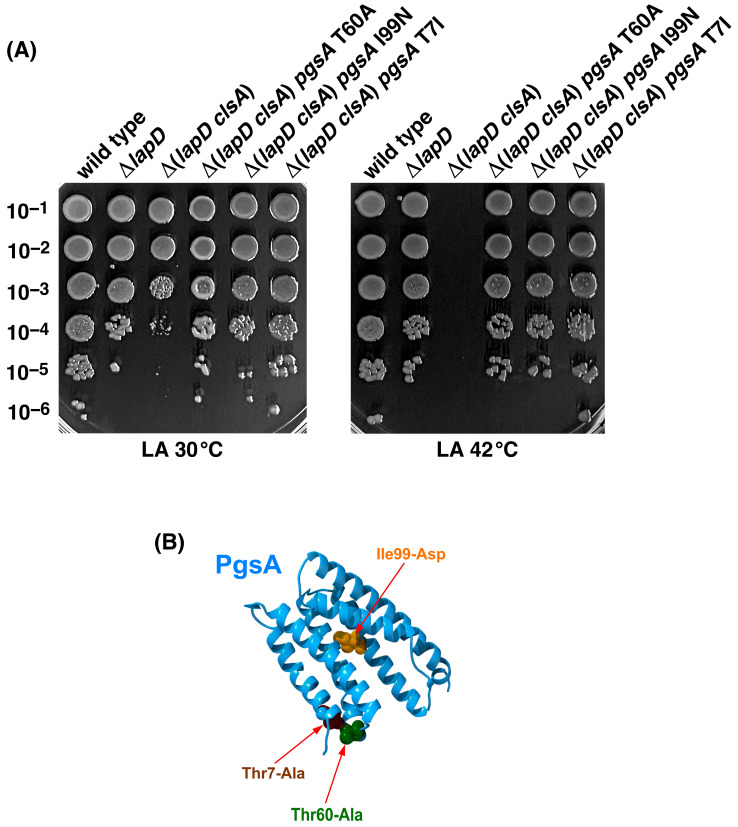 Figure 3
Figure 3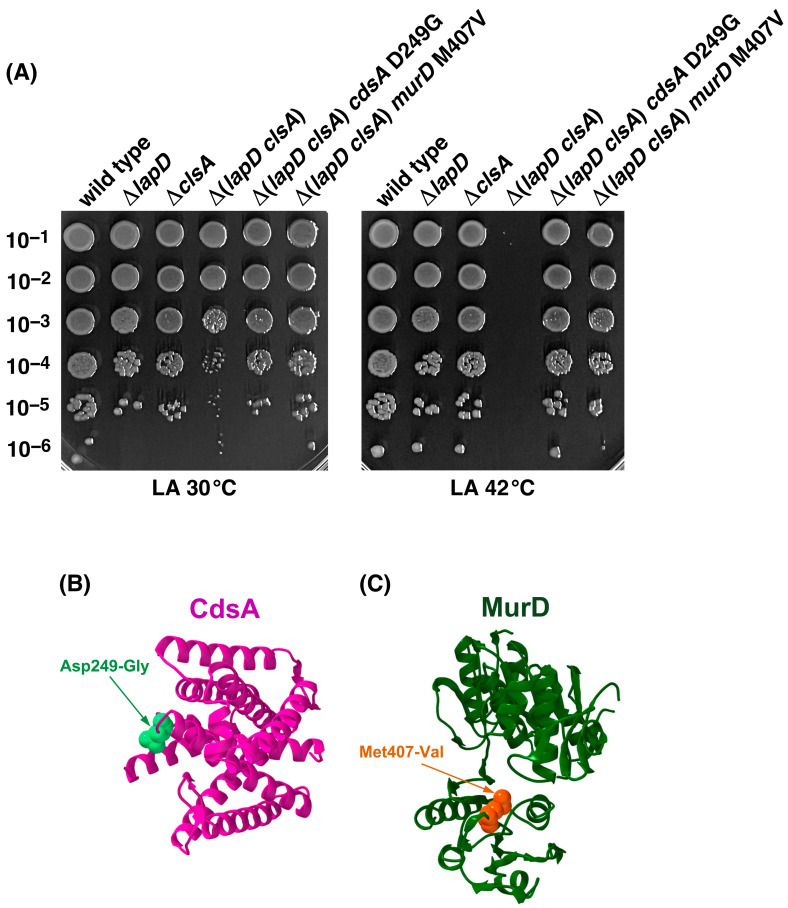 Figure 4
Figure 4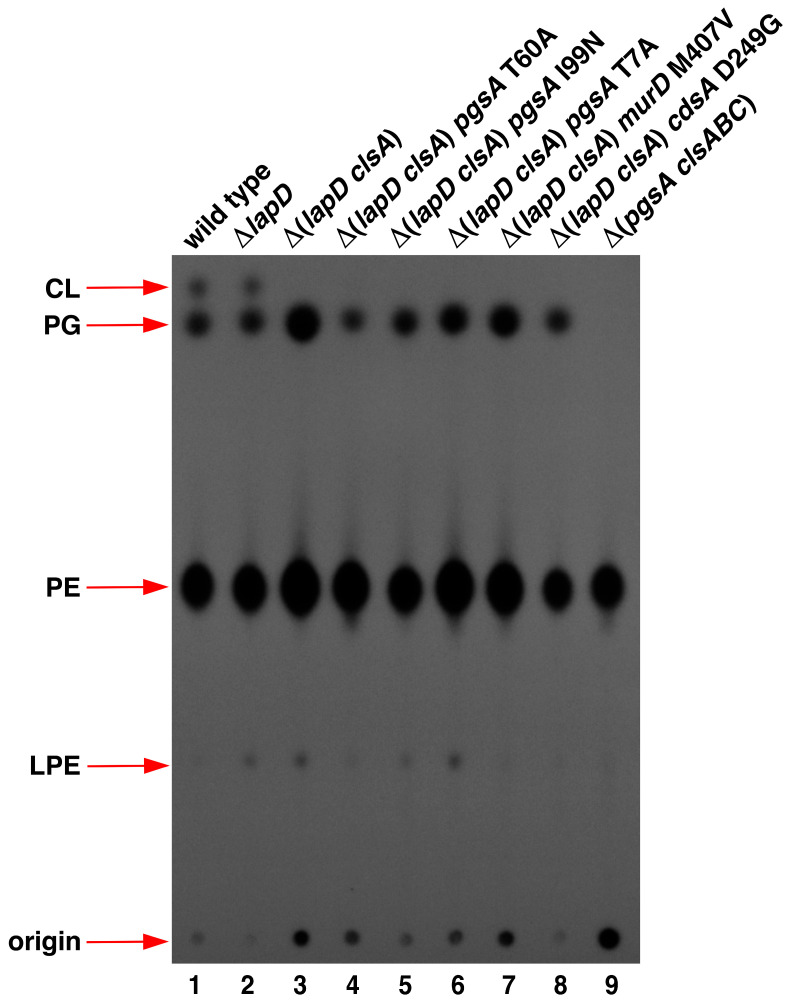 Figure 5
Figure 5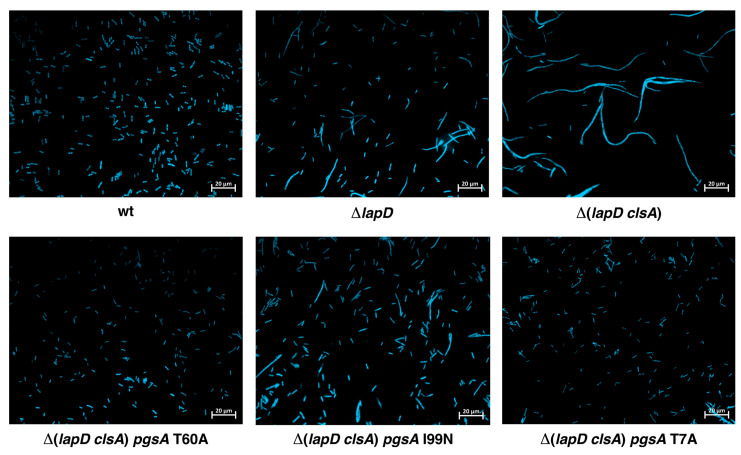 Figure 6
Figure 6 Figure 7
Figure 7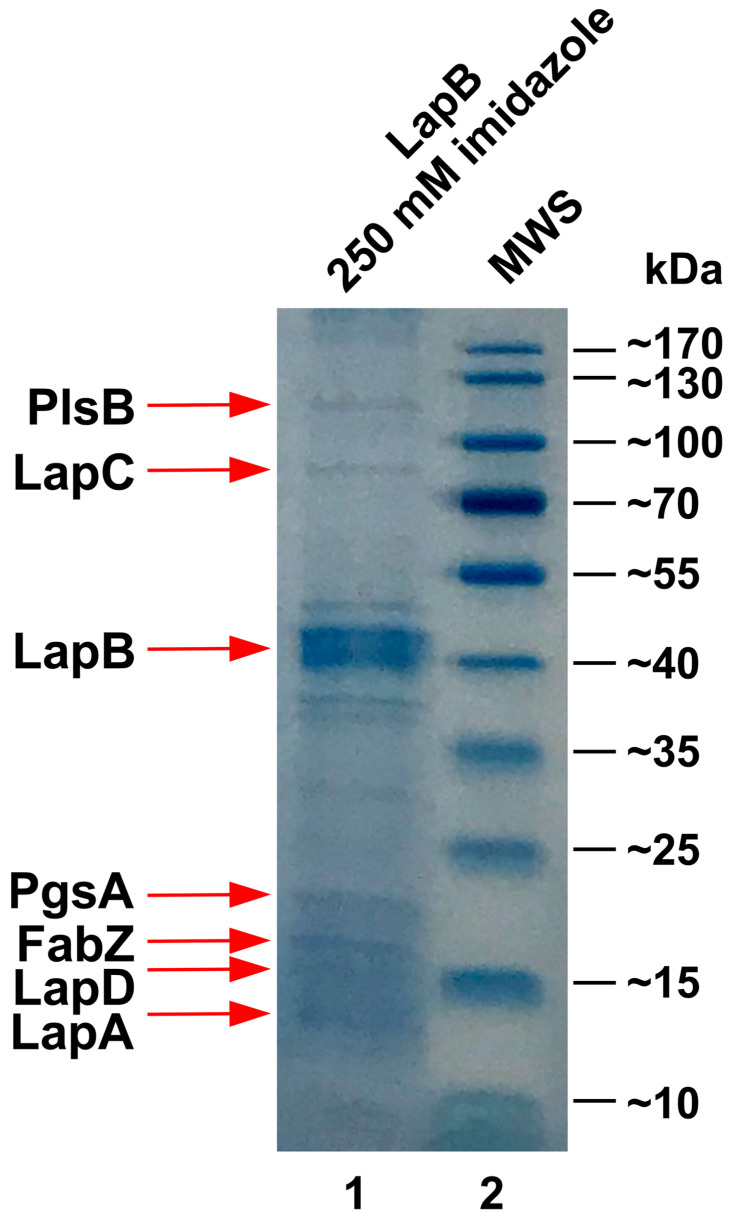 Figure 8
Figure 8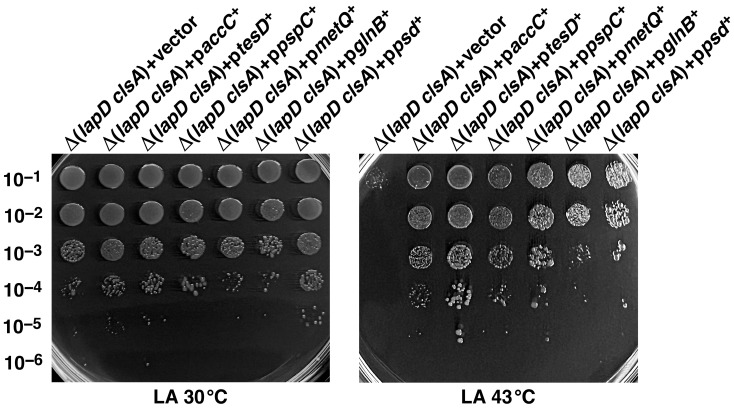 Figure 9
Figure 9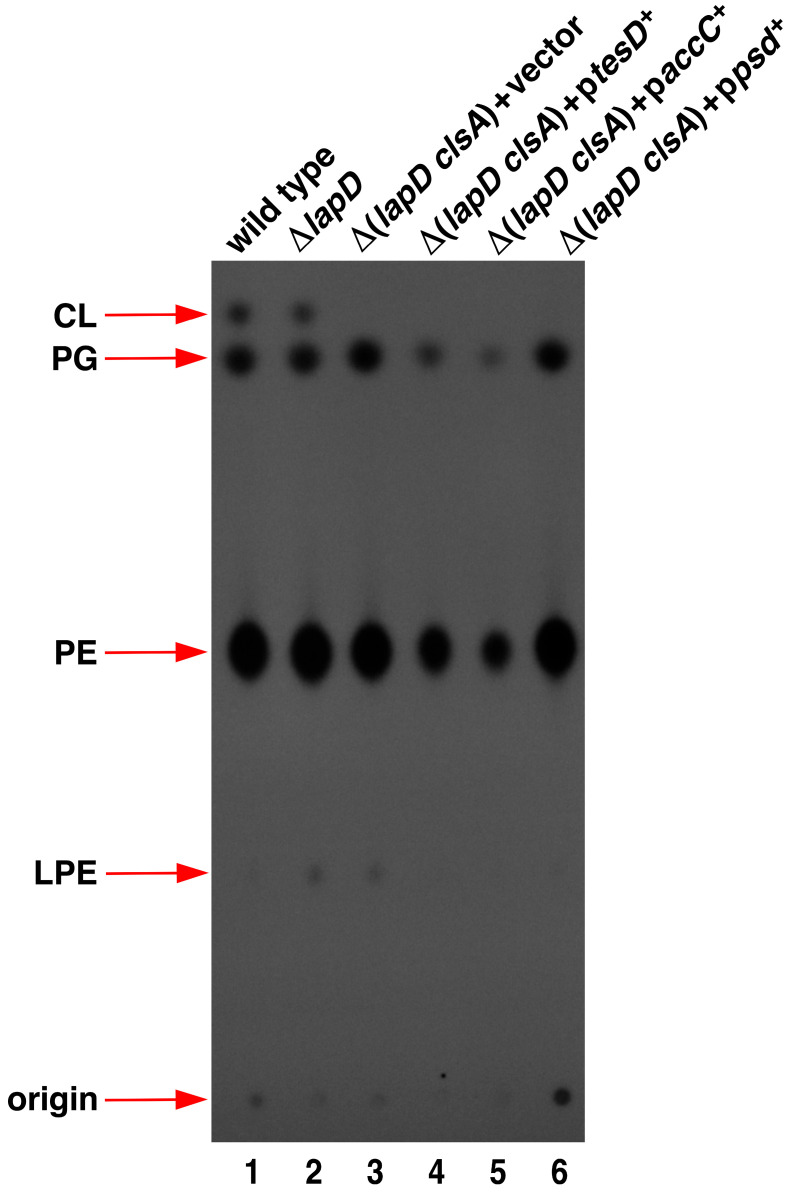 Figure 10
Figure 10 Figure 11
Figure 11- —National Science Center (NCN)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Genetics and Biotechnology · Microbial Metabolic Engineering and Bioproduction · Vibrio bacteria research studies
1. Introduction
The cell envelope of Gram-negative bacteria, including Escherichia coli, contains two distinct membranes, an inner (IM) and an outer (OM) membrane, separated by the periplasm, which includes a thin layer of peptidoglycan. The defining feature of the OM of Gram-negative bacteria is its asymmetry due to the presence of phospholipids (PLs) in the inner leaflet and the location of lipopolysaccharide (LPS) in the outer leaflet [1]. LPS constitutes the major component of the OM, and its essentiality is due to its role in providing a permeability barrier and structural integrity of the OM [2]. Similarly, PLs are integral in regulating membrane integrity and its fluidity [3]. Both of these components of OM are essential for bacterial viability, and their amounts are tightly regulated, as these two essential components use the same β-hydroxymyristoyl-ACP as a common metabolic precursor [4,5]. Hence, any imbalance between LPS and PLs leads to a loss of bacterial viability. Thus, bacteria, such as E. coli, hold a constant ratio of 0.15:1.0 for these two essential components, respectively [6]. However, the mechanisms by which bacteria maintain homeostatic control are poorly understood. One of the most studied regulatory controls that has received recent attention is the regulated turnover of LpxC, which catalyzes the first committed step in LPS biosynthesis [7]. This involves the positive regulation of LpxC proteolysis by the FtsH-LapB complex and its negative control by LapC [5,8,9]. PLs are derived from fatty acids (FA). Bacteria, such as E. coli, have devised controls for the biosynthesis of new fatty acids and the modification of the structure of existing fatty acids [3]. Similarly, LPS can undergo structural non-stoichiometric modifications in its lipid A and core parts, which impart resistance to cationic antibiotics [10,11,12]. Together, these processes allow bacteria to adjust LPS amounts according to their demand and control membrane viscosity to match environmental requirements. Another player, LapD, has recently been implicated in the regulation of LPS and fatty acid balance, although the precise mechanism of its involvement remains unknown [13,14].
In E. coli LPS biosynthesis, a tetraacylated lipid A precursor is first synthesized via a pathway that requires six essential enzymes [2,15]. Two Kdo residues are added to lipid IV_A_ by the WaaA enzyme, followed by the addition of secondary lauroyl and myristoyl chains, resulting in the synthesis of hexaacylated lipid A. Additional sugars are added to this key intermediate, leading to the LPS core formation. In smooth bacteria, a distal O-antigen is further attached after flipping LPS across the IM on the periplasmic side [15]. The synthesis of hexaacylated LPS is crucial because LPS that is either tetraacylated or pentaacylated is poorly translocated to the OM [16,17]. Furthermore, the absence of myristoyl acyltransferase LpxM causes synthetic lethality when either LPS or PL synthesis is compromised [13,14,18,19]. The fatty acid biosynthesis pathway in E. coli is of the type FASII system that comprises four steps using both acetyl coenzyme A (acetyl-CoA) and malonyl-ACP as starter units and malonyl-ACP as the extender unit [3]. In the initiation of FA biosynthesis, acetyl-CoA carboxylase (ACC) catalyzes the first committed step in fatty acid biosynthesis: the conversion of acetyl-CoA to malonyl-CoA [20,21]. Each turn of the FA biosynthesis cycle adds two carbon atoms to the growing acyl chain. The long-chain acyl-ACP thioesters generated during FA synthesis serve as acyl donors for PLs synthesis [22]. Phosphatidic acid (PA) serves as a universal precursor for membrane PL formation [23]. In E. coli, PA is generated by the acylation of sn-glycerol-3-phosphate (G3P) to 1-acyl-G3P, followed by a second acylation, requiring PlsB and PlsC acyltransferases, respectively [23,24]. PA is then converted to the central precursor of all phospholipids, cytidine diphosphate-diacylglycerol (CDP-DAG), by CDP-DAG synthase encoded by the cdsA gene (Figure 1). The primary PLs in E. coli are phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and cardiolipin (CL). PE and PG are essential for bacterial viability, whereas CL is dispensable. CDP-DAG is positioned at a branch point in PL biosynthesis, where it reacts with sn-glycerol 3-phosphate to form PG or with L-serine to synthesize phosphatidylserine (Figure 1) [25]. CL is produced via the condensation of two PG molecules or a single PE and PG molecule [22]. The balance of zwitterionic (PE) and acidic phospholipids (PG and CL) in E. coli stems from the continued synthesis of PG by the PG phosphate synthase–phosphatase (PgsA–PgpA/B) system, which is coupled with the tight regulation of PE synthesis through the PssA–Psd pathway [3] (Figure 1).
E. coli has three CL synthases, with ClsA being the major contributor for producing CL. Although nonessential for bacterial viability, CL plays a crucial role in maintaining structural integrity, cell size, and regulating the activity of various proteins, such as the polar localization of ProP, assembly of LacY, and protein transport through the Sec apparatus [26,27,28]. More importantly, it was shown that CL presence is essential for the viability of strains lacking either WaaA Kdo transferase or LpxM myristoyl acyltransferase [19]. As suppressors that overcome the synthetic lethality of Δ(clsA lpxM) mapped to MsbA LPS flippase, it was proposed that CL participates in LPS translocation [18,19].
Biochemical characterization of LapD revealed that it co-purifies with enzymes involved in PL, FA, and LPS biosynthesis. Although LapD is non-essential, its absence confers synthetic lethality when lipid A is underacylated or when LPS is composed only of Kdo_2_-lipid A [13,14]. Δ(lapD lpxM) bacteria were found to contain elevated amounts of acidic phospholipids and significant alterations in FA composition, supporting a role of LapD in regulation of FA composition. Consistent with this hypothesis, suppressor mutations that overcome the lethal phenotype of Δ(lapD lpxM) were found to map to genes encoding various subunits of the ACC complex, which restored PL amounts. However, the absence of LapD also causes retention of LPS in the IM, and suppressors that relieve the conditional lethal phenotype of Δ(lapD waaC) bacteria map to either genes involved in LPS assembly in the OM (lptD) or in the nlpI gene, whose product regulates proteolysis of peptidoglycan hydrolase [14]. Thus, LapD is an enigmatic protein, and significant gaps remain in our understanding of the pleiotropic phenotypes associated with its absence.
In this study, we showed that Δ(lapD clsA) bacteria exhibited conditional lethality. Thus, we used this synthetic lethal phenotype of Δ(lapD clsA) to isolate single-copy chromosomal and multicopy suppressors that overcame this lethality and addressed their cell morphology defects and PL/fatty acid composition (Figure 2). Most single-copy extragenic suppressors mapped to genes involved in PL biosynthesis. These include single amino acid exchanges in the pgsA gene, whose product catalyzes the condensation of CDP-DAG with G3P to form the intermediate PG phosphate (PGP), which is then dephosphorylated by Pgp phosphatases to produce PG (Figure 1). Another set of suppressors carried a mutation mapping to the essential cdsA gene, which encodes CDP-DAG synthetase that catalyzes the synthesis of CDP-DAG [29]. Mutations in pgsA and cdsA genes reduced PL levels, which are otherwise elevated in Δ(lapD clsA) bacteria, and restored the fatty acid composition. Identification and characterization of multicopy suppressors revealed that overexpression of genes (accC and glnB), whose products inhibit fatty acid biosynthesis at the level of the ACC enzyme, or the tesD gene encoding a putative thioesterase, which causes a decrease in the overall PL content, overcomes the lethality of Δ(lapD clsA) bacteria. We further showed that PgsA co-purifies with LapB/D complex and that Δ(lapD clsA) bacteria exhibit gross alterations in cellular morphology, which are suppressed by loss-of-function mutations in the pgsA gene. These genetic and biochemical studies revealed that the lethality of Δ(lapD clsA) is due to the toxic accumulation of PL moieties and that LapD acts as a nodal point in the homeostatic control of FA and LPS biosynthesis.
2. Results
2.1. Suppressor-Free Δ(lapD clsA) Bacteria Exhibit Conditional Synthetic Lethality
The function of LapD is not well understood but has been implicated in cell envelope-related functions, including a role in maintaining a balance between LPS and PLs. Thus, we undertook several experiments to address the role of LapD when PL biosynthesis is impaired. One of the non-essential PL components is CL, but its cellular requirement is not well understood. The major enzyme required for cardiolipin formation under exponential growth conditions is ClsA [30,31]. Individually, lapD and clsA genes are dispensable for bacterial viability under laboratory growth conditions. However, as shown earlier, the clsA gene becomes essential when LPS is underacylated and exhibits a sick phenotype in the absence of LapD [13]. We reconstructed Δ(lapD clsA) strains in the W3110 background, which has been extensively used for LPS and PL analysis, because some other background strains, such as BW25113, carry a non-functional fabR gene [32]. Thus, reciprocal bacteriophage P1-mediated transductions were carried out in the wild type and its isogenic derivatives lacking either lapD or clsA genes with the vector alone or with a covering plasmid. Parallel transductants were plated on LA plates at 30, 37, and 42 °C. The results from these experiments revealed that ΔlapD and ΔclsA can be introduced into the wild type at the same frequency at the three temperatures. However, Δ(lapD clsA) or reciprocal Δ(clsA lapD) viable transductants could be obtained at 30 °C with or without covering the plasmid, although the transductants were obtained at a lower frequency (Table 1). These transductants exhibited a small colony morphology. At 42 °C, no viable Δ(lapD clsA) and Δ(clsA lapD) transductants could be obtained without covering the plasmid (Table 1). At 37 °C, transductants could be obtained but at a highly reduced frequency. These results allow us to conclude that the Δ(lapD clsA) combination is synthetically lethal at 42 °C and, although viable at 30 °C, exhibits a sick phenotype.
2.2. Suppressors That Allow Δ(lapD clsA) Bacterial Growth at Elevated Temperatures Map to Phospholipid Biosynthetic Genes pgsA, cdsA, and Those Involved in Peptidoglycan Biosynthesis
To address the molecular basis of the conditional synthetic lethality of Δ(lapD clsA) bacteria, extragenic suppressors that allow growth at 42 °C were sought by plating cultures at elevated temperatures. Temperature-resistant (Ts^+^) survivors were obtained at a frequency of approximately 10^−8^. Ts^+^ independently obtained suppressors were marked with the Tn10 transposon, as described previously [14]. Linked Ts^+^ mutations were verified by retransducing the suppressor mutation into the parental Δ(lapD clsA) strain SR25209. Fourteen independent suppressors with transposon linked >90% were retained after verification. To map the suppressor mutations, Tn10 insertions were recombined onto cosmids. The identity of the suppressor mutation was determined by sequencing the PCR products of the complementing cosmids. These mutations could be grouped into three complementation groups, with 10 out of 14 linked to the uvrC gene. The second complementation group of three strains with the suppressor mutation was linked to the skp gene. The third complementation group had Tn10 mapping to the mraZ gene, located next to the gene cluster carrying genes whose products are involved in cell division and peptidoglycan biosynthesis. DNA sequencing of the region next to uvrC, skp, and mraZ genes revealed single amino acid exchanges in the essential pgsA, cdsA, and murD genes, respectively. These results further showed that five independently isolated suppressors had the suppressor mutation in the pgsA gene encoding phosphatidylglycerophosphate synthase, causing alteration of aa T60. Four out of five had a single amino acid exchange of T60A (ACC to GCC) in the pgsA gene (Table 2). Another suppressor-carrying strain had a single amino acid alteration, T60P, in the coding region of the pgsA gene. Significantly, the T60A mutation results in the alteration of aa 60, which is the same residue with a substitution to be the first reported and very well characterized pgsA3 mutation [33,34,35]. However, the original pgsA has an exchange of T60P, as opposed to our T60A substitution, which was found in four out of five independently obtained Ts^+^ suppressors. Significantly, it is encouraging that we also obtained one suppressor mutation that is identical to the original pgsA3 mutation. The pgsA3 mutation in the pgsA gene lacks the potential to synthesize phosphatidylglycerolphosphate, a precursor of PG [36]. The original pgsA3 was identified as a Ts^+^ suppressor mutation of the (pssA-1 clsA) strain [36]. Similarly to the pgsA3 mutation, pgsA T60A was found to be recessive, as it could be complemented by a cosmid clone carrying the pgsA gene. Further DNA sequence analysis revealed that three strains carried a single amino acid substitution of T7A (ACG to GCG) in the coding region of the pgsA gene, and the two remaining strains carried a single amino acid substitution of I99N at aa position 99 due to ATC to AAC nucleotide alteration (Table 2). All three amino acid residues, T60, T7, and I99, are highly conserved in PgsA orthologs, and substitution of T60 has been shown to cause a loss of enzymatic activity (Figure 3).
The complementation group linked to the skp gene, comprising three Ts^+^ suppressor-bearing strains, had a single amino acid substitution at aa position 249 due to GAC to GCC alteration in the coding region of the cdsA gene, resulting in the D249G substitution. The cdsA gene encodes CDP-diglyceride synthetase, which catalyzes the synthesis of CDP-diacylglycerol (CDP-DAG) using CTP and phosphatidic acid (PA) as substrates [29,37]. As CDP-DAG serves as a precursor of all PLs in E. coli, we expect that the suppressor mutation could act by reducing the overall PL amounts. DNA sequencing analysis of the adjoining regions of the Tn10 mutation revealed that the last Ts suppressor of Δ(lapD clsA) carries a suppressor mutation in the essential murD gene due to the exchange of M407V (ATG to GTG). The murD gene encodes UDP-N-acetylmuramoyl-L-alanine:D-glutamate ligase, which catalyzes the addition of the second amino acid to the peptide moiety of the monomer unit of peptidoglycan [38]. The amino acid residue 407 is located in the conserved C-terminal domain, which could be important for the stability and specificity of the MurD enzyme [38].
2.3. Evaluation of Growth Properties of Suppressors That Overcome Synthetic Lethality of Δ(lapD clsA) Bacteria at Elevated Temperatures
As the majority of single-copy suppressor mutations mapped to the pgsA gene that overcame the synthetic lethality of Δ(lapD clsA) bacteria, we estimated their growth properties using a spot-dilution assay at 30 and 42 °C. The wild type, ΔlapD, and isogenic strains with suppressor mutations in the pgsA gene showed normal growth at 30 and 42 °C. However, Δ(lapD clsA) bacteria exhibited a 10-fold reduction in colony-forming ability, even at 30 °C, and lethality at 42 °C (Figure 3). Thus, the Δ(lapD clsA) combination conferred a sick phenotype at 30 °C, with a severe reduction in the colony size. Furthermore, Δ(lapD clsA) bacteria are unable to grow at elevated temperatures and is synthetically lethal at 42 °C. In parallel, we quantified the growth properties of Δ(lapD clsA) derivatives with suppressor mutations mapping to cdsA and murD genes at 30 and 42 °C. The spot-dilution assay again revealed the sick phenotype of Δ(lapD clsA) bacteria at 30 °C and the lethal phenotype at 42 °C. Importantly, suppressor mutations in either cdsA or murD genes restored nearly wild-type-like growth at 42 °C, validating their isolation (Figure 4).
2.4. Restoration of Phosphatidylglycerol Levels in Δ(lapD clsA) Bacteria by Suppressor Mutations in PgsA
To address the molecular basis of the synthetic lethality of Δ(lapD clsA) bacteria, the PL composition of panels of strains with and without suppressor mutations in pgsA, cdsA, and murD genes was analyzed using thin-layer chromatography (TLC). PL were extracted as a control from the strain RU857, which cannot synthesize cardiolipin (CL) and phosphatidylglycerol (PG) species and serves as an internal marker for various PL species. The cultures were labeled with inorganic phosphate, and PL was extracted as described previously [14]. Equivalent amounts of total radiolabeled PL were applied after adjusting for the total protein content. PL species were visualized using phosphorimaging analysis and quantified by densitometry. Densitometric quantification of different PL species revealed that the wild-type strain contained approximately 74.30% ± 3.9 PE, followed by 19.15% ± 0.8 PG and 6.54% ± 0.5 CL (Figure 5, lane 1). These values are consistent with those of the three known PL species in E. coli [39]. No major differences in the composition of PL species were observed between the extracts obtained from wild-type and ΔlapD bacteria, except for the minor accumulation of lysophosphatidylethanolamine (LPE) in ΔlapD and in some of its derivatives (Figure 5, lane 2). However, most significantly, Δ(lapD clsA) contained approximately a 4.2-fold increase in PG and a 1.5-fold increase in PE compared to the wild type (Figure 5, lane 3). Interestingly, the isogenic strain Δ(lapD clsA) with the pgsA T60A suppressor mutation repressed this vast accumulation of PG, reducing it by 5.9-fold compared to the wild-type level (Figure 5, lane 4). Other suppressor mutations, pgsA I99N and pgsA T7A, also reduced the elevated accumulation of PG amounts (Figure 5, lanes 5 and 6), although not as dramatically as observed with the pgsA T60A suppressor mutation. The suppressor mutation pgsA I99N reduced PG levels by nearly 1.8-fold compared to those in the parental Δ(lapD clsA) strain. The suppressor mutation cdsA D249G reduced elevated PE levels by more than 15% compared to that in the wild type (Figure 5, lane 8). However, no major differences were observed with the suppressor mutation in the murD gene, which is not expected to directly regulate PL amounts and would likely only regulate PGN. Thus, we can conclude that suppressor mutations in either pgsA or cdsA genes relieve the lethality of Δ(lapD clsA) by reducing the toxic accumulation of elevated PG and PE levels.
2.5. Δ(lapD clsA) Bacteria Exhibit Gross Alterations in Fatty Acid Composition, Which Are Suppressed by Mutations in PgsA
It is well established that fatty acid and PL synthesis are intricately linked. In the above sections, we showed that Δ(lapD clsA) bacteria have significantly altered amounts of PL species, particularly with hyperelevated amounts of PG. Thus, we further analyzed the fatty acid composition of Δ(lapD clsA) bacteria and their isogenic suppressors that overcome their lethal phenotype. Cultures were grown in LB medium, and free fatty acids (FFA) were extracted from an isogenic panel of strains grown at 28 and 37 °C and analyzed using gas chromatography (GC).
GC quantification of FFA extracted from cultures grown at 28 °C revealed that most significantly Δ(lapD clsA) had approximately 20% less unsaturated fatty acid (UFA) species 18:1 cis-vaccenic acid than the wild type (Table 3). The most robust suppressor mutation in Δ(lapD clsA) pgsA T60A reversed this composition, leading to an increase in more than 23% in 18:1 cis-vaccenic acid content compared to that in the wild type. Interestingly, the suppressor mutations pgsA T7A and cdsA D249G restored 18:1 levels to nearly wild-type levels (Table 3). Furthermore, the Δ(lapD clsA) strain accumulated 3-fold more amounts of saturated fatty acid 18:0 as compared to the parental strain, which was partly reversed by the pgsA T60A mutation. Significantly, Δ(lapD clsA) pgsA T60A bacteria accumulated only 58% myristic acid (14:0). Another major difference is an approximately 30% decrease compared to the wild type in the amount of 16:1 palmitoleic acid UFA when pgsA T60A is present in Δ(lapD clsA) bacteria at 28 °C.
Analysis of GC data from FFA extracted from bacteria grown at 37 °C further reinforces the results from 28 °C that the severe growth defects of Δ(lapD clsA) bacteria are due to gross alterations in the fatty acid composition. The amount of UFA cis-vaccenic acid 18:1 species was even more reduced, being approximately 64% of that in the wild type (Table 3). This was reversed by suppressor mutations in pgsA and cdsA genes. The suppressor mutation pgsA T60A caused more than a 32% increase in the 18:1 UFA content compared to the wild type. Notably, Δ(lapD clsA) bacteria exhibited an increased presence of 12:0 and 14:0 saturated fatty acids, which were repressed by some of the suppressor mutations, particularly for myristic acid (14:0). Taken together, the results from the GC experiments allow us to conclude that the absence of CL in ΔlapD bacteria results in large-scale changes in the biosynthesis of fatty acids, some of which are reversed by suppressor mutations in pgsA and cdsA genes. Thus, these results can explain the molecular basis of the synthetic lethality of the Δ(lapD clsA) combination and provide a rationale for the isolation of suppressor mutations in the PL biosynthetic pathway. These results are consistent with the known bacterial requirement to maintain a balance between saturated and unsaturated fatty acids, which is critical for membrane fluidity, and in this process, LapD and ClsA functions are critical.
2.6. Δ(lapD clsA) Bacteria Have Severe Defects in Cell Morphology, Which Are Suppressed by Mutations in PgsA
Fatty acid composition is known to control the cell length [40]. Increased fatty acid synthesis can lead to abnormal cell expansion, resulting in filamentous cell morphology. ΔlapD bacteria also exhibit cell morphology defects but are viable under normal laboratory growth conditions [14]. As Δ(lapD clsA) bacteria exhibit a sick phenotype even at 30 °C and have elevated PG levels with alterations in fatty acid composition, the cell morphology of such bacteria was examined in parallel with isogenic parental strains and derivatives with single amino acid substitutions that overcome their lethal phenotype. Exponentially grown bacteria were analyzed using fluorescence microscopy after staining with DAPI. ΔlapD bacteria exhibited a high proportion of filamentous cells. However, this phenotype was exacerbated in Δ(lapD clsA) bacteria (Figure 6). Most of the cells of Δ(lapD clsA) bacteria exhibited abnormally long cellular morphology. Quite significantly, suppressor mutations in the pgsA gene suppressed this severe long filamentous phenotype (Figure 6). The more robust suppressor mutation pgsA T60A virtually restored wild-type- like cell morphology. Notably, pgsA T7A also effectively suppressed the filamentous morphology of Δ(lapD clsA) bacteria (Figure 6). These data are consistent with the restoration of PL levels and fatty acid composition by such suppressor mutations. Taken together, the bacterial synthetic lethality of Δ(lapD clsA) and the sick phenotypes under permissive growth conditions are due to changes in fatty acid composition concomitant with severe cell morphology defects.
2.7. Suppressors of Δ(lapD clsA) Bacteria Do Not Alter LpxC Levels
As PL and LPS biosynthesis utilize (R)-3-hydroxymyristate-ACP as a common metabolic precursor, it is possible that altered PL and fatty acid synthesis due to suppressor mutations in pgsA and cdsA might cause disturbances in LPS synthesis at the level of LpxC. LpxC is a key enzyme involved in regulating the balance between the LPS and PL biosynthetic pathways [4,5]. Thus, we examined LpxC levels using Western blotting. Isogenic cultures of wild type, ΔlapD, ΔclsA, Δ(lapD clsA), and its derivatives with suppressor mutations in pgsA, cdsA, and murD genes were grown under permissive growth conditions. As a control, cell lysates were prepared from isogenic ΔftsH sfhC21 bacteria that are known to have elevated levels of LpxC [4]. Equivalent amounts of proteins were transferred by Western blotting and immunoblotted with anti-LpxC antibodies. Estimation of LpxC levels revealed that none of the suppressor mutations in the pgsA gene caused any significant alteration in its amount (Figure 7, panel A). As expected, ftsH mutant bacteria showed increased LpxC levels. No impact of the clsA gene deletion was observed. Similarly, no significant changes in LpxC levels of Δ(lapD clsA) derivatives with the suppressor mutation in either cdsA or murD genes were observed (Figure 7, panel B). These results allow us to conclude that the single-copy extragenic suppressor mutation in either pgsA or cdsA or murD genes that overcome the lethal phenotype of the Δ(lapD clsA) combination do not operate by changing LpxC levels and are unlikely to impact LPS biosynthesis.
2.8. PgsA Co-Purifies with the Essential LapB Protein
Previously, we showed that the inner membrane essential protein LapD co-purifies with several proteins involved in LPS biosynthesis or its assembly (LapC, LapA, WaaC, and LapD) and the initiation of PL synthesis (FabZ) [5,8,13]. Hence, it was proposed that, in addition to regulating LpxC stability via interaction with FtsH, LapB could act as a scaffold for recruiting various enzymes in PL and LPS to coordinate their synthesis using its conserved tetratricopeptide repeats (TPR) [5]. Thus, we wondered whether any of the proteins examined in this study are also part of the LapB interactome. To achieve this, pull-down experiments were performed using the affinity purification of C-terminally His-tagged LapB, as described previously [5]. His-tagged LapB co-eluted with several proteins (Figure 8). MALDI-TOF analyses of co-purifying proteins revealed that the LapB complex, in addition to including known interacting partners such as LapA, LapC, LapD, and FabZ, also includes PgsA and PlsB (Figure 8). PlsB glycerol-3-phosphate acyltransferase catalyzes the first committed step in phospholipid biosynthesis [23]. Co-purification of PgsA and PlsB suggests that LapB-interacting partners are dedicated to PL and LPS biosynthesis.
2.9. Multicopy Suppressors of Δ(lapD clsA) Identify Factors That Can Inhibit Fatty Acid Biosynthesis or Regulate Cell Envelope Homeostasis
To further understand the physiological limiting factors that cause the lethality of Δ(lapD clsA) bacteria, multicopy suppressor analysis was performed. Two genomic plasmid DNA libraries were used. One of these was based on the ordered ASKA genomic library of cloned ORFs, where the expression of individual genes is under the control of an inducible, tightly regulated P_T5_-lac promoter [41]. Transformants of Δ(lapD clsA) with this library were selected at 42 °C in the presence of 75 μM IPTG. The second library was generated using the mini-Mu system [42]. In this case, the first deletion derivatives of clsA and lapD genes were transduced into the p15A mini-Mu lysogenic strain, and lysates prepared from such strain were used to generate plasmid libraries. Transductants in the Δ(lapD clsA) background were plated to isolate Ts^+^ colonies. Plasmid DNAs from such Ts^+^ strains were isolated, verified by retransformation, and used to identify the minimal coding sequence by DNA sequencing that could restore the growth of Δ(lapD clsA) bacteria. The usage of mini-Mu-based libraries was necessitated to prevent recloning of lapD and clsA genes, which was the case with the ASKA library. These multicopy suppressors identified a set of genes involved in fatty acid/PL synthesis or regulation of cell envelope-related functions (Table 4).
To validate these results, the suppression ability was analyzed using a spot-dilution assay of Δ(lapD clsA) derivatives carrying these genes expressed from the P_T5_-lac promoter (Figure 9). These results revealed a variable degree of restoration of bacterial growth at 43 °C in the presence of 75 μM IPTG, although all the analyzed strains performed better than the parental strain with the vector alone. The most robust suppression was observed with the gene encoding putative thioesterase tesD, followed by pspC and accC genes when mildly overexpressed (Figure 9). Noticeable restoration of bacterial growth at 43 °C was also observed when glnB, psd, and metQ genes were overexpressed.
The products of some genes can inhibit fatty acid biosynthesis or affect PL biosynthesis. The most prominent of these was the repeated cloning of the accC gene, which encodes the biotin carboxyl carrier protein, one of the four subunits of the ACC complex [21]. AccC catalyzes the first step of the acetyl-CoA carboxylase reaction mediated by the ACC enzyme. It has been demonstrated that for the enzymatic activity of the ACC complex, all four subunits must be maintained in stoichiometric amounts, and any imbalance can inhibit the ACC enzymatic activity [14]. These results are consistent with the demonstration of a reduction in the amount of PL when one of the subunits of the ACC complex is overproduced or mutated [14]. Another interesting multicopy suppressor identified is the glnB gene (Table 4). The glnB gene encodes the PII-1 protein, which is involved in the regulation of nitrogen metabolism by controlling the activity of glutamine synthetase [43,44,45]. However, more importantly, in the context of this study, GlnB also acts as a regulator of fatty acid synthesis via its interaction with the biotin carboxylase/biotin carboxyl carrier protein (BC-BCCP) subcomplex of the ACC enzyme [46]. GlnB interacts with BC-BCCP to form a ternary complex that inhibits the ACC enzyme activity [46]. Another gene whose overexpression rescues the lethality of Δ(lapD clsA) is the psd gene, encoding phosphatidylserine decarboxylase. Psd catalyzes the formation of the most abundant PL species, phosphatidylethanolamine. The isolation of PspC as a dosage-dependent suppressor can be rationalized, as it has been shown to be a substrate of FtsH protease [47]. MetQ is a lipoprotein and its overproduction could cause diversion of PG pools that can overcome toxic accumulation of PG and thereby overcome lethality. Thus, multicopy suppressor analysis further reinforces our results that LapD and ClsA absence leads to dysfunctional fatty acid/PL biosynthesis, causing an increase in PG amounts. Currently, the precise function of TesD is unknown; however, its predicted polypeptide has a characteristic hot-dog fold observed in the family of thioesterases and could act by hydrolyzing acyl chains.
2.10. Overexpression of accC and tesD Suppresses Elevated Accumulation of PL Species in Δ(lapD clsA) Bacteria
To address the molecular basis of the restoration of growth of Δ(lapD clsA) bacteria by overexpression of various multicopy suppressor-encoding genes, PL analysis was undertaken using TLC. We specifically analyzed those, which are predicted to inhibit fatty acid biosynthesis and alter PL composition. Thus, isogenic cultures of derivatives of Δ(lapD clsA) with the vector alone or expressing plasmid-born accC, psd, and tesD were labelled with inorganic phosphate in the presence of 75 μM IPTG to induce gene expression. Samples from such cultures were used to extract PLs and analyzed for their composition using TLC. As is evident, Δ(lapD clsA) had more than 33% elevation of PG amounts compared to the parental wild type, which was significantly suppressed by overexpression of either tesD or accC gene (Figure 10, lanes 3 vs. 4 and lanes 3 vs. 5). Densitometric quantification of PL species when the tesD gene was overexpressed showed PG to be approximately 47% and PE nearly 63% compared to the parental wild type, which was taken as 100%. An even more severe reduction in PG and PE amounts was observed when the accC gene was overexpressed. Thus, the PG amounts were approximately 24% and PE only 46% compared to the parental wild type upon accC overexpression. As mentioned above, overproduction of any of the subunits of the ACC complex can disturb the balance in the stoichiometry of various components, leading to the inhibition of ACC enzymatic activity that catalyzes the first committed step in the initiation of fatty acid synthesis. If TesD is proven to be a thioesterase, it could impact by releasing acyl chains by hydrolysis that can reduce PL synthesis. No significant alterations were observed with overexpression of the psd gene, consistent with the observation of homeostatic control of fatty acid biosynthetic enzymes and the lack of changes in PL composition due to the amplification of Psd enzymes [48].
2.11. Overexpression of pspC Elevates LpxC Levels
One of the multicopy suppressors that restores the growth of Δ(lapD clsA) at high temperatures is the pspC gene. PspC is a positive regulator of the psp operon and has been suggested to be a substrate of FtsH protease [47]. The FtsH-LapB complex catalyzes the proteolysis of LpxC. Thus, we rationalized that excess of PspC could titrate FtsH, which could potentially stabilize LpxC. To test this hypothesis, we examined LpxC levels using immunoblotting. Total cell lysates were prepared from isogenic wild type, Δ(lapD clsA), and its derivative expressing the pspC gene under the control of the inducible P_T5_-lac promoter and analyzed by Western blotting. Estimation of LpxC levels revealed that overexpression of the pspC gene led to an increase in LpxC amounts compared to those observed in the parental wild type and its Δ(lapD clsA) derivative (Figure 11). These results provide a rationale explanation for the isolation of pspC gene as a multicopy suppressor of the Ts phenotype of Δ(lapD clsA) bacteria by stabilization of LpxC.
3. Discussion
The outer membrane of Gram-negative bacteria is primarily composed of lipopolysaccharides and phospholipids. In E. coli, FASII generates two products, acyl-ACP and β-hydroxyacyl-ACP, which are components of LPS and PLs biosynthesis. Acyl-ACPs are used by PlsB and PlsC enzymes to generate phosphatidic acid, which is a precursor of all cytoplasmic membrane phospho- and glycolipids [3]. β-Hydroxyacyl-ACP molecules serve as substrates for the acyltransferases catalyzing the initial steps in the biosynthesis of the essential conserved lipid A component of LPS. We previously showed that LapD is one of the factors that links the LPS and PL biosynthetic pathways, as it co-purifies with various components of PL and LPS biosynthesis [13]. Genetic and biochemical studies have shown that although LapD is non-essential for bacterial growth, it becomes indispensable when the lipid A is underacylated (absence of LpxM myristoyl acyltransferase) or when LPS is composed of only Kdo_2_-lipid A due to the lack of heptosyltransferase WaaC [13,14]. We showed that suppressors that bypass the lethality of Δ(lapD lpxM) and Δ(lapD waaC) map to either various subunits of the essential enzyme acetyl-CoA carboxylase transferase or components of LPS transport and peptidoglycan biosynthesis [14]. Here, we show that LapD also becomes conditionally essential in the absence of the major cardiolipin biosynthesis pathway. CLs are the only PLs that are non-essential for bacterial viability. Thus, it is intriguing that cardiolipins are required for bacterial viability in the absence of LapD. However, CL synthesis has been shown to be required for viability of bacteria lacking LpxM [18,19]. Although the Δ(lapD clsA) mutational combination has been shown to confer lethality, its molecular basis has not been addressed [13,49]. In this study, we showed that Δ(lapD clsA) bacteria exhibited severe morphological defects. We used this conditional synthetic lethal phenotype to isolate extragenic chromosomal and multicopy suppressors to investigate its molecular basis.
Characterization of these suppressors showed that the majority of single-copy suppressor mutations mapped to the pgsA gene, encoding phosphatidylglycerophosphate synthase, whose product is required for PG synthesis (Figure 1). PG synthesis occurs via the condensation of CDP-DAG with G3P mediated by PgsA to form the intermediate PG phosphate, which is then dephosphorylated by Pgp phosphatase to form PG [50]. Another set of suppressor mutations was mapped to the cdsA gene encoding CDP-diglyceride synthetase. CDP-DAG is the central precursor of all PL in E. coli. Interestingly, the most frequent single amino acid exchange observed in the Ts^+^ suppressors of Δ(lapD clsA) was in the amino acid residue Thr 60 in PgsA. Alteration of this highly conserved amino acid residue impairs PgsA activity [36,51]. To our knowledge, this is the first study to identify suppressor mutations in the pgsA gene in the absence of LapD. Analysis of the PL content of Δ(lapD clsA) bacteria revealed elevated levels of PG accumulation, which can contribute to the toxicity and severe defects in cell division observed in these bacteria. Importantly, loss-of-function pgsA suppressor mutations restored the wild-type-like cell morphology and significantly reduced PG abundance. Notably, suppressor mutations in the pgsA gene were shown to repress highly elevated PL levels, particularly in the presence of the PgsA T60A amino acid exchange. Thus, ΔlapD bacteria are sensitized due to an imbalance in either PL composition, or underacylation of lipid A, or when LPS is composed of only Kdo_2_-lipid A. While extragenic suppressors of Δ(lpxM clsA) were mapped to the msbA gene encoding LPS flippase [16], suppressors of Δ(lapD clsA) were mapped to PL biosynthetic genes, implying that CLs have a dual function. Thus, CLs can assist MsbA in LPS translocation and cooperate with LapD to maintain a balanced fatty acid composition.
Acidic phospholipids, including CL, localize to cell poles and interact with key players such as RodZ [52,53]. The absence of CL in ΔlapD could be one of the reasons for the severe cell morphology defects in Δ(lapD clsA) bacteria. Consistent with this notion, rodZ has been shown to be essential in the absence of LapD, although no explanation for this lethality has been provided [54]. Furthermore, it is well established that E. coli size is dependent on fatty acid/PL levels [40,55,56]. The suppression of cell morphology defects of Δ(lapD clsA) bacteria by loss-of-function mutations in the pgsA gene is consistent with the prominent role of anionic PLs in cell morphology. Anionic lipids have been shown to be required for the assembly of the Z-ring and for DNA replication [57,58,59]. In this study, we observed an abnormally high accumulation of PG in Δ(lapD clsA) bacteria, which was relieved by pgsA suppressor mutations. Consistent with these results, we previously showed restoration of normal cell morphology when fatty acid biosynthesis was repressed by overexpression of either the tesA’ thioesterase gene, induction of acpP, or inhibition of ACC complex activity [14].
Interestingly, gross alterations in Δ(lapD clsA) bacteria were observed in FFA composition with an increased presence of saturated vs. unsaturated fatty acids, which was particularly reflected by a 3-fold increase in 18:0 and a 20–40% decrease in the presence of cis-vaccenic acid (18:1). Increased accumulation of cis-vaccenic unsaturated fatty acid can lead to a decrease in LPS biosynthesis, which can reset the balance between LPS and PL amounts, as observed in Δ(lapD lpxM) bacteria [14]. Significantly, cis-vaccenic acid amounts were restored in the presence of suppressor mutations in either cdsA or pgsA genes. Overall, these results imply that the Δ(lapD clsA) lethal phenotype stems from alterations in the fatty acid and PL composition. Another interesting suppressor mutation was mapped to the essential murD gene, which connects PGN synthesis and LapD function, consistent with our earlier findings [14].
In support of the results revealing excess PG and changes in fatty acid composition, the multicopy suppressor approach revealed that reducing fatty acid synthesis by inhibiting acetyl-CoA carboxylase activity, which catalyzes the rate-limiting step in the initiation of fatty acid synthesis, can overcome the lethality at elevated temperatures. This approach revealed that overexpression of either accC, encoding the subunit of the ACC enzyme, or glnB effectively restored the growth of Δ(lapD clsA) bacteria. The ACC complex has four subunits, and all components are held in stoichiometric amounts [60]. Overexpression of any of the subunits reduces PL and fatty acid content by decreasing the ACC enzyme activity [14]. We showed a significant decrease in PG molecules, which are otherwise elevated in Δ(lapD clsA) bacteria when the AccC subunit is overproduced, providing a genetic and biochemical link to the reasons for the lethality and mode of growth restoration of Δ(lapD clsA) bacteria. Another multicopy suppressor is encoded by the glnB gene, whose product belongs to the PII signal transduction family and plays a key role in nitrogen metabolism [43,44,45]. However, GlnB proteins from Proteobacteria have also been shown to interact with the biotin carboxyl carrier protein (BCCP) of acetyl-CoA carboxylase (ACC) [46,61]. In E. coli, this GlnB interaction reduces the k_cat_ of acetyl-CoA carboxylation [46]. This overproduction of GlnB should result in the reduced ACC enzyme activity, which can provide a rational explanation for its role as a dosage-dependent suppressor. The isolation of metQ as a multicopy suppressor is interesting because the encoded protein is a predicted lipoprotein that is part of the methionine transport system. Prosite annotation suggests lipidation at amino acid position 23, which could be a PG moiety. MetQ is included in the TIGRFAM lipoprotein TIGR00363/NlpI family (http://ca.expasy.org/prosite (accessed on 9 October 2025)). PspC, whose overproduction also suppresses the Ts phenotype, acts as a positive regulator of the psp operon [62]. We speculate that overproduction of MetQ could titrate out PG molecules, which are present in toxic amounts in Δ(lapD clsA) bacteria. However, MetQ is not well characterized, and further studies are required to validate its lipidation and its impact on PL pools. However, it has parallels, as the absence of the most abundant lipoprotein, Lpp, allows the growth of pgsA mutant bacteria [63]. The rationale for Δlpp to relive the lethality of pgsA mutants is based on the known PG attachment to this highly abundant lipoprotein, which can lead to the availability of PG molecules that are very limiting in pgsA mutant bacteria [64]. The isolation of pspC as a multicopy suppressor is interesting and can be explained by the observed stabilization of LpxC upon its overexpression in Δ(lapD clsA) bacteria. This is consistent with PspC being a substrate of FtsH [47]. We explain these results because titration of FtsH by excess of PspC can lead to the stabilization of LpxC. This can reset the LPS vs. PL balance, which is disturbed in Δ(lapD clsA) bacteria. These results are further supported by our previous findings showing that LpxC stabilization can overcome the growth defects of ΔlapD bacteria [13]. Thus, increasing either the stability of LpxC or repressing PL synthesis can overcome the lethality of Δ(lapD clsA) bacteria, positioning LapD as the link between LPS and FA synthesis. Another possibility of PspC-mediated suppression is the association of CL with Psp proteins in polar membrane regions, which can affect interactions with the RodZ/MreB cytoskeleton complex and Tat-dependent protein translocation [65]. It is possible that PspC is limiting in Δ(lapD clsA) bacteria, which can impact the extracytoplasmic stress response, which is elevated in the absence of LapD [13,14].
Similarly, we can explain the isolation of putative thioesterase TesD, as its overproduction reduced PL synthesis and restored growth at elevated temperatures. We have not yet biochemically established the thioesterase activity of TesD, and this awaits confirmation. However, TesD has a characteristic hot-dog fold, which is present in the thioesterase superfamily [66]. Thioesterases typically catalyze the cleavage of thioester bonds in a wide range of activated fatty acyl coenzyme A (CoA) substrates, acyl carrier proteins (ACPs), and other cellular molecules [66]. Importantly, it has been shown that thioesterase overproduction can alter the degree of saturation of the membrane lipids in E. coli [67]. It has been shown that the biosynthetic coupling between fatty acid and phospholipid syntheses could be disrupted by the high-level expression of thioesterases [68]. The isolation of tesD is also supported by our earlier results with overexpression in the cytoplasm of tesA’ lacking its signal sequence, which effectively suppressed PL accumulation in ΔlapD derivatives [14]. Isolation of psd as a multicopy suppressor is also logical, as its product encodes phosphatidylserine decarboxylase catalyzing the synthesis of PE.
Thus, based on our results of the synthetic lethality of the concomitant absence of LapD and CL, we can conclude that they together constitute essential components required to maintain the homeostasis of essential cell envelope components. In such bacteria, several key components, particularly the composition of PLs, fatty acids, and cellular morphology, are severely affected. Thus, Δ(lapD clsA) bacteria accumulated toxic amounts of PG species of PLs, causing gross alterations in the fatty acid composition, which resulted in defects in cellular morphology. Our results show that lethality can be overcome by reducing PG synthesis, as shown by the preponderance of suppressor mutations that relieve this lethality mapping to the pgsA gene. Furthermore, Δ(lapD clsA) bacteria accumulated less unsaturated fatty acids, particularly cis-vaccenic acid, with a significant increase in saturated fatty acids. This imbalance can cause alterations in the regulation of membrane fluidity and hence, Δ(lapD clsA) bacteria cannot withstand temperatures above 37 °C, which contributes to the synthetic lethal phenotype. Suppressor mutations in the pgsA gene, which are recessive, not only reduced the hyperelevated toxic accumulation of PG but also corrected cellular morphological defects. These findings were supported by the isolation of multicopy suppressors, whose overexpression can inhibit the initiation of fatty acid at the level of the acetyl coenzyme A carboxylase enzyme. Notably, we identified a new putative thioesterase, TesD, whose overproduction effectively prevents the synthetic lethality of Δ(lapD clsA) bacteria and significantly reduces the accumulation of elevated PL species. Currently, efforts are underway to establish the thioesterase activity of TesD and its specificity for the acyl chain length. Thus, we can conclude that the synthetic lethality of Δ(lapD clsA) is due to the increased toxic accumulation of PG species and an altered ratio of saturated and unsaturated fatty acids. Hence, consistent with our present results and previous findings, LapD is a multitasking protein that connects LPS, PL, and PGN synthesis, and cardiolipins play a pivotal role in this process. This is further supported by the finding that PgsA is part of the large protein complex of LapB/LapD, which can lead to the coordination of LPS and PL biosynthetic pathways.
4. Materials and Methods
4.1. Bacterial Strains, Plasmids and Media
The bacterial strains and plasmids used in this study are described in Table 5.
Luria–Bertani (LB) broth and agar (LA) (Difco, Franklin Lakes, NJ, USA) were prepared as described earlier [75]. As per the experimental requirements, the media were supplemented with ampicillin (100 μg/mL), kanamycin (50 μg/mL), tetracycline (10 μg/mL), or chloramphenicol (20 or 30 μg/mL). The expression of various genes in the pCA24N expression vector was induced by adding 75 μM IPTG to the growth medium.
4.2. Strain Construction and Isolation of Extragenic Chromosomal Suppressors
All strains used in this study were derived from W3110, serving as the parental wild-type strain, unless otherwise stated. The construction of a non-polar antibiotic-free deletion derivative of lapD SR25204 in W3110 has been previously described [14]. Bacteriophage P1-mediated transductions were performed as previously described [76]. The ΔlapD derivative SR25204 served further as a recipient to construct the Δ(lapD clsA) derivative SR25209 by bacteriophage P1-mediated transduction. To construct this strain, the ΔclsA non-polar deletion was first constructed by replacing the coding sequence of the clsA gene by the aph cassette from the pKD13 plasmid using the λ recombinase system, as described [17]. The aph antibiotic cassette was removed by transformation with pCP20 plasmid DNA, which expresses a site-specific recombination flippase [72]. To construct Δ(lapD clsA), bacteriophage P1-mediated transductions were performed in reciprocal order and plated on LA agar plates at 30 °C, which was found to be a permissive condition for such derivatives. As a control, each recipient in the transduction experiments was transformed with a covering plasmid expressing either lapD or clsA genes. To isolate extragenic chromosomal suppressor mutants, cultures of several independently obtained suppressor-free Δ(lapD clsA) transductants grown at 30 °C were plated at 42 °C. The Ts^+^ properties were verified by re-streaking. Cultures from these Δ(lapD clsA) Ts^+^ isolates, 65 putative suppressor-containing strains, were retained. Suppressor mutations were marked with mini-Tn10 Tet [77], and verified that the Tn10-linked suppressor mutation breeds true. To identify the suppressor mutation, linked Tn10 mutations were recombined onto single-copy cosmid clones using a previously described DNA library [14,74]. To identify the candidate gene with a suppressor mutation, DNA from recombinant cosmids was subcloned, selecting for Tn10 Tet and Amp markers or by inverse PCR using chromosomal DNA to identify Tn10 junctions, as described [78]. DNA from the recombinant plasmid was used to sequence the Tn10 ends and flanking regions using inverse PCR, as previously described [8,78]. This allowed us to place 14 suppressor mutations in three complementation groups. The same cosmid DNAs were then used to sequence the candidate genes using specific oligonucleotides.
4.3. Identification of Multicopy Suppressors, Whose Overexpression Overcomes Lethal Phenotype at 42 °C
DNA from plasmid pools obtained from the ordered genomic library cloned in pCA24N carrying all predicted ORFs of E. coli (ASKA collection) [41] was used to transform the Δ(lapD clsA) strain SR25209, selecting directly for Ts^+^ survivors at 42 °C in the presence of 75 μM IPTG. Cultures for Ts^+^ colonies were grown to isolate plasmid DNAs, which were used to retransform the Δ(lapD clsA) strain SR25209 for the validation of growth restoration at 42 °C. The identity of the cloned gene, whose overexpression conferred suppressing ability, was obtained by DNA sequencing. However, this method gave a lot of background due to the recloning of lapD and clsA genes, although several bona fide multicopy suppressors were also isolated. To overcome this drawback, the mini-Mu in vivo cloning approach was used [42,70]. To achieve this, a Δ(lapD clsA) derivative was constructed in a derivative with Muc ts62 Mud5005, resulting in the construction of SR23769. Mini-Mu lysates of SR23769 were prepared by thermal induction and used to transduce plasmids into Δ(lapD clsA) derivative SR25209, selecting for Ts^+^ colonies at 42 °C. Plasmid DNAs from such Ts^+^ derivatives were subcloned into a p15A-based medium copy plasmid vector pMBL18 [73] to identify the minimal coding region that could suppress the lethality of Δ(lapD clsA) at 42 °C. Plasmids that bred true in suppressing ability were used for DNA sequencing.
4.4. Isolation and Analysis of 32P-Labeled Phospholipid Species
Overnight cultures of isogenic strains of wild type, ΔlapD, Δ(lapD clsA), and its derivatives with suppressor mutations in pgsA, cdsA, and murD were grown in LB medium under permissive growth conditions at 30 °C. The cultures were diluted, adjusted to an OD_595_ of 0.05, and grown for another 45 min and labelled with 2.5 μCi/mL of ^32^P_i_ in 2.5 mL of LB medium until an OD_595_ of 1.0. For the growth of strains Δ(lapD clsA) derivatives carrying multicopy suppressors, during the labelling period, 75 μM IPTG was added for the expression of P_T5_-lac promoter-inducible accC, psd, and tesD genes. Cultures were harvested by centrifugation at 4300× g for 10 min. The pellets were dispersed in water. Phospholipids were extracted using the procedure described by Bligh and Dyer, and Ames [79,80]. The PLs were collected from the lower organic phase and dried under a stream of nitrogen. Dried samples containing phospholipids were reconstituted in 50 μL of a 2:1 chloroform/methanol mixture. Before the extraction of PLs, an aliquot of the samples was used to measure the total protein concentration using a Pierce BCA protein assay kit (ThermoScientific, Warsaw, Poland). Equivalent amounts (10,000 cpm/lane) of ^32^P-labeled phospholipids after adjusting for protein concentration were separated on TLC Silica gel 60 F254, aluminum sheets, 20 cm × 20 cm plates (Merck, Mississauga, ON, Canada). Lipids were separated using a solvent system, as described [14]. The TLC plates were dried and exposed to CL-Xposure^Tm^ Film (ThermoScientific, Rockford, IL, USA) or exposed to a PhosphorImager screen to visualize and quantify the labeled PLs using the Amersham Typhoon Biomolecular Imager (Cytiva, Uppsala, Sweden). Densitometry analysis of the autoradiograms was performed using the ImageJ software (rsb.info.nih.gov). Three biological replicates were used for quantification.
4.5. Isolation of Fatty Acids and Their Analysis Using Gas Chromatography
Isogenic bacterial cultures were grown at 28 and 37 °C as described for phospholipid extraction, without adding either IPTG or ^32^P_i_. Fifteen mL cultures at an OD_595_ of 1.0 were harvested by centrifugation at 4300× g for 10 min, and the pellets were frozen. The pellets were thoroughly resuspended in 2.5 mL of H_2_O with vortexing, followed by the addition of 5 μL of 10 mg/mL heptadecanoic acid (C17:0) (Merck, Warsaw, Poland) dissolved in ethanol, as an internal standard. Free fatty acids were extracted as per the procedure described [14,81]. Fatty acid methyl esters were obtained by treating the dried extract with 0.5 mL of 1.25 M HCl in methanol and incubating at 50 °C for 15 h. Fatty acids were extracted twice in 500 μL of hexane. FFA were analyzed by gas chromatography using a Shimadzu GC-2010 Pro (Shimadzu, Kyoto, Japan) with a 30 m × 0.25 mm capillary column under the experimental conditions described [14]. One µL of each sample was injected using a 1:100 split ratio of the helium carrier gas. For calibration, a fatty acid methyl ester mixture C8-C22 (CRM18920 Merck, Warsaw, Poland) was used to establish the retention time of various peaks corresponding to different fatty acids. Each experiment was repeated with three independent biological repeats. The data were analyzed using the software provided by Shimadzu (Kyoto, Japan).
4.6. Estimation of LpxC Amounts by Immunoblotting
Isogenic strains of wild type, Δ(lapD clsA), and its derivatives with either chromosomal extragenic suppressor mutation or carrying plasmid born multicopy suppressors were routinely grown at 30 °C in 5 mL LB medium up to an OD_595_ of 1.0. The cultures were harvested by centrifugation at 7000× g for 10 min. The cell pellets were resuspended in SDS lysis buffer. As an internal control, cell lysates were prepared from the ΔftsH sfhC21 derivative grown under similar conditions. Protein concentrations were measured using the Pierce BCA protein assay kit (ThermoScientific, Warsaw, Poland). An equivalent amount of protein was applied to a 12% SDS-PAGE for resolution. After electrophoresis, the proteins were blotted to a PVDF membrane. Membranes were immunoblotted with LpxC-specific antibodies and revealed using a chemiluminescence kit (ThermoScientific, Warsaw, Poland) according to the manufacturer’s instructions.
4.7. Examination of Cellular Morphology and Measurement of Bacterial Growth
For quantification of bacterial growth, isogenic bacterial cultures were grown overnight in LB medium at 30 °C. Exponentially grown cultures were adjusted to an OD_595_ of 0.1, serially diluted, and bacterial growth was measured using spot-dilution assay as described [82]. To examine cellular morphology, cultures of wild type, ΔlapD, Δ(lapD clsA), and its three isogenic derivatives with independent chromosomal suppressor mutations in the pgsA gene were grown in LB medium. Overnight cultures were diluted 1:100 and allowed to grow to an OD_595_ of 0.6 under permissive conditions at 30 °C in LB medium. Aliquots of cultures were centrifuged at 4300× g for 5 min and incubated for 10 min in TBS supplemented with 10 µg/mL of 4′,6-diamidino-2-phenylindole (DAPI) stain. Samples were immobilized on agarose pads, and cell morphology was examined using epifluorescence and differential interference contrast (DIC) microscopy, as described [14]. Cellular morphology was observed using a Zeiss apotome microscope (Carl Zeiss, Jena, Germany).
4.8. Purification of LapB
The production of C-terminal His_6_-tagged LapB was induced with 0.3 mM IPTG, using expression system as described [5]. Inner membrane proteins were extracted in binding buffer containing 50 mM NaH_2_PO_4_, 300 mM NaCl, 10 mM imidazole (buffer A) supplemented with 1% octyl-β-D-glucoside. After centrifugation, the supernatant was purified by FPLC (AKTA Pure, Cytiva, Warsaw, Poland) using a 1 mL HisTrap FF column. The column was washed with a binding buffer containing 20 mM imidazole to remove non-tagged proteins. Proteins were eluted with buffer A using a step gradient ranging from 50, 100, 250, and 500 mM imidazole and analyzed on 12% SDS-PAGE.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nikaido H. Molecular basis of bacterial outer membrane permeability revisited Microbiol. Mol. Biol. Rev.20036759365610.1128/MMBR.67.4.593-656.200314665678 PMC 309051 · doi ↗ · pubmed ↗
- 2Klein G. Wieczorek A. Szuster M. Raina S. Checkpoints that regulate balanced biosynthesis of lipopolysaccharide and its essentiality in Escherichia coli Int. J. Mol. Sci.20222318910.3390/ijms 23010189 PMC 874569235008618 · doi ↗ · pubmed ↗
- 3Zhang Y.M. Rock C.O. Membrane lipid homeostasis in bacteria Nat. Rev. Microbiol.2008622223310.1038/nrmicro 183918264115 · doi ↗ · pubmed ↗
- 4Ogura T. Inoue K. Tatsuta T. Suzaki T. Karata K. Young K. Su L.H. Fierke C.A. Jackman J.E. Raetz C.R.H. Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease Fts H (Hfl B) in Escherichia coli Mol. Microbiol.19993183384410.1046/j.1365-2958.1999.01221.x 10048027 · doi ↗ · pubmed ↗
- 5Klein G. Kobylak N. Lindner B. Stupak A. Raina S. Assembly of lipopolysaccharide in Escherichia coli requires the essential Lap B heat shock protein J. Biol. Chem.2014289148291485310.1074/jbc.M 113.53949424722986 PMC 4031536 · doi ↗ · pubmed ↗
- 6Mohan S. Kelly T.M. Eveland S.S. Raetz C.R. Anderson M.S. An Escherichia coli gene (fab Z) encoding (3R)-hydroxymyristoyl acyl carrier protein dehydrase. Relation to fab A and suppression of mutations in lipid A biosynthesis J. Biol. Chem.1994269328963290310.1016/S 0021-9258(20)30075-27806516 · doi ↗ · pubmed ↗
- 7Sorensen P.G. Lutkenhaus J. Young K. Eveland S.S. Anderson M.S. Raetz C.R.H. Regulation of UDP-3-O-[R-3-hydroxymyristoyl]-N-acetylglucosamine deacetylase in Escherichia coli. The second enzymatic step of lipid A biosynthesis J. Biol. Chem.1996271258982590510.1074/jbc.271.42.258988824222 · doi ↗ · pubmed ↗
- 8Biernacka D. Gorzelak P. Klein G. Raina S. Regulation of the first committed step in lipopolysaccharide biosynthesis catalyzed by Lpx C requires the essential protein Lap C (Yej M) and Hsl VU protease Int. J. Mol. Sci.202021908810.3390/ijms 2123908833260377 PMC 7730581 · doi ↗ · pubmed ↗