Enantioselective α,α-Chlorofluorination of Sulfoxonium Ylides
Lucas G. Furniel, Kauê C. Capellaro, Viktor S. Câmara, Marcio Hayashi, Radell Echemendía, Camila B. Pinto, Ana B. A. M. Salata, Jackson A. L. Filho, Leandro W. Hantao, Javier Ellena, Antonio C. B. Burtoloso

TL;DR
This paper reports a new method for creating chiral gem-dihalogenated compounds with high efficiency and enantioselectivity.
Contribution
The paper introduces enantioselective α,α-chlorofluorination using chiral aminosulfoxonium ylides and fluorinating reagents from cinchona alkaloids.
Findings
25 examples of gem-dihalogenated compounds were synthesized with up to 90% yield.
Enantioselectivity reached up to 96:4 er using chiral fluorinating reagents.
The method includes bromochlorination and is derived from commercially available cinchona alkaloids.
Abstract
The first examples of enantioselective α,α-chlorofluorination of α-carbonylsulfoxonium ylides are described. Herein, two modes of reaction are explored, using chiral aminosulfoxonium ylides and a chiral fluorinating reagent, prepared in situ from commercially available cinchona alkaloids. Using these approaches, 25 examples of gem-dihalogenated compounds (including bromochlorination) were obtained in good yields (up to 90%) and enantioselectivity (up to 96:4 er).
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
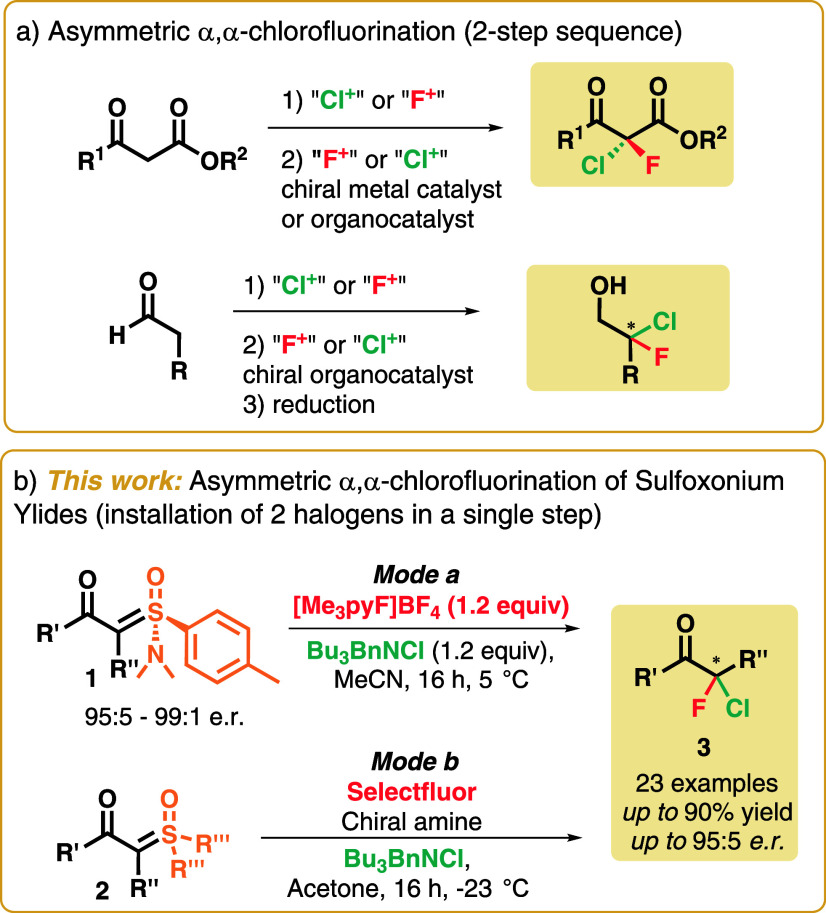 Figure 1
Figure 1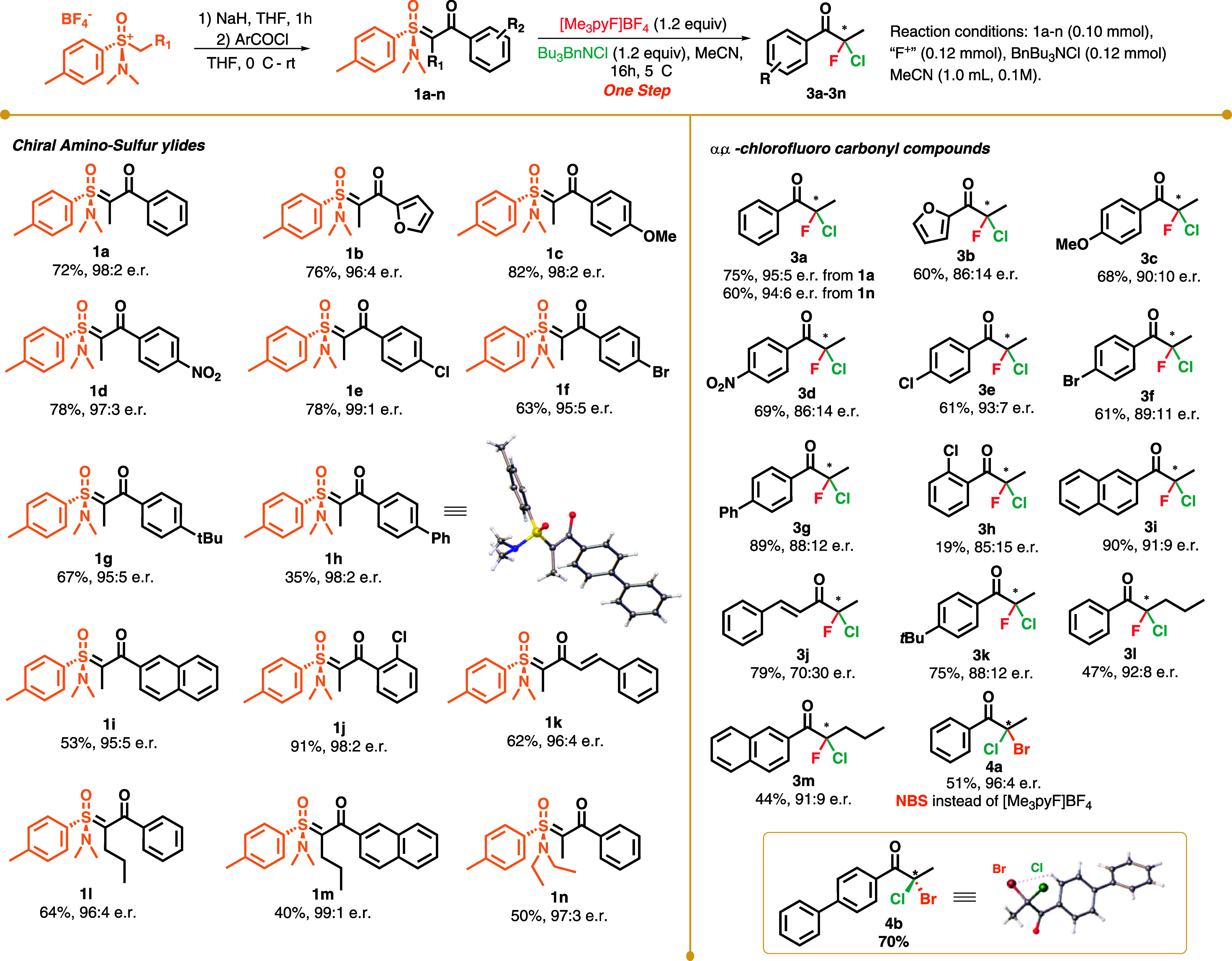 Figure 2
Figure 2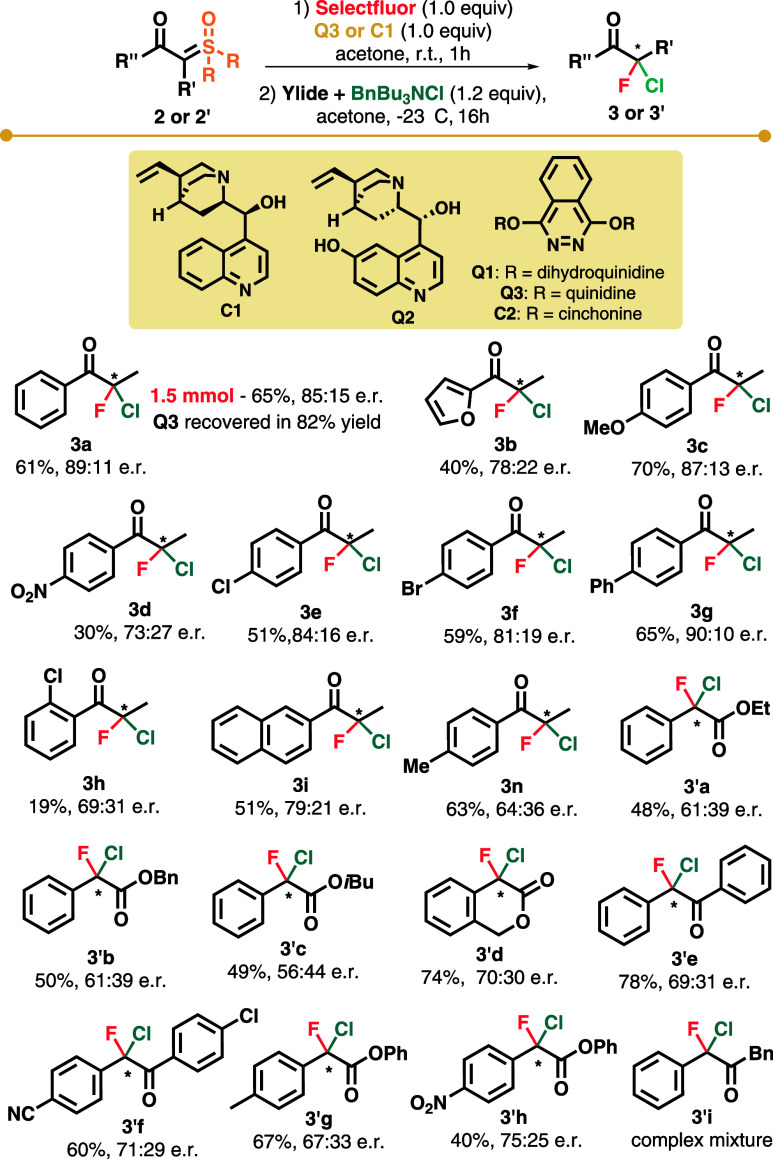 Figure 3
Figure 3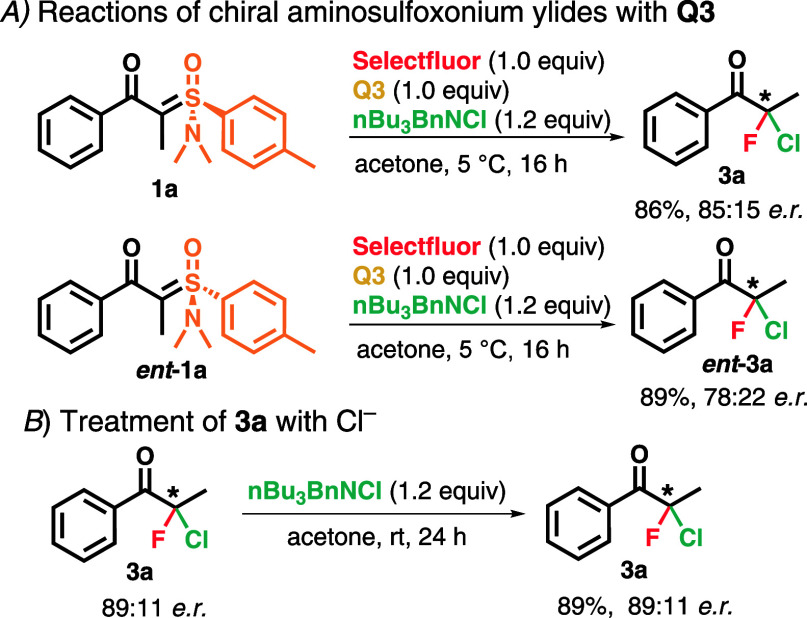 Figure 4
Figure 4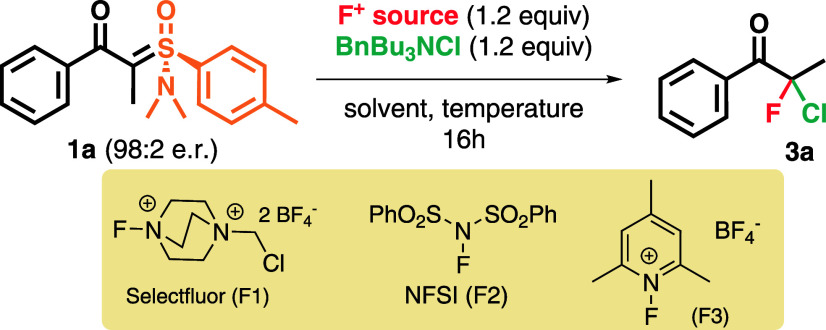 Figure 5
Figure 5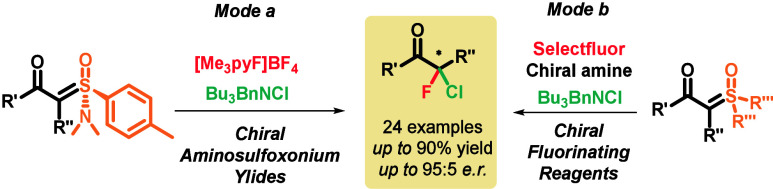 Figure 6
Figure 6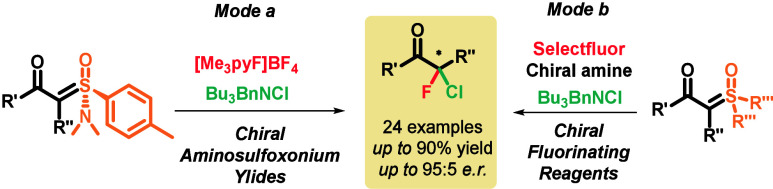 Figure 7
Figure 7- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Coordena??o de Aperfei?oamento de Pessoal de N?vel Superior10.13039/501100002322
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFluorine in Organic Chemistry · Synthesis and Characterization of Pyrroles · Vanadium and Halogenation Chemistry
Fluorine substituents can significantly alter the pK a of neighboring groups and affect properties such as dipole moments, metabolic stability, lipophilicity, and bioavailability. ?,? These characteristics have made fluorine ubiquitous in agrochemicals and pharmaceuticals. ?−? ? Due to these important characteristics and the rarity of naturally occurring fluorine-containing molecules,? several methodologies for C–F bond formation have been developed over the past 30 years. ?−? ? ? ? Despite numerous advances in this field, fewer than 1% of pharmaceuticals containing fluorine atoms have a C–F bond at chiral nonracemic carbon centers, primarily due to the inherent challenges in constructing these centers.? To address this limitation, various strategies have been employed to enable the asymmetric formation of C–F bonds with high enantioselectivity, with particular focus on α-fluorocarbonyl compounds. ?−? ? ? ? ? ? ? ? ?
In contrast to these advances in asymmetric α-monofluorination, reports of enantioselective α,α-chlorofluorination are rare. ?−? ? ? ? ? ? ? ? ? ? To the best of our knowledge, only 10 studies have been published to date. In each of these cases, two separate steps are required: one for the installation of the first halogen in a racemic fashion followed by asymmetric monohalogenation of the previously halogenated substrate (Schemea). These strategies are limited to aldehydes or dicarbonyl compounds and, in many cases, suffer from low yields in the first halogenation step due to the unwanted formation of difluorinated or dichlorinated side products. These limitations make the development of a one-step asymmetric α,α-chlorofluorination reaction a significant challenge in organic synthesis. Once formed, α,α-chlorofluorocarbonyl compounds are highly versatile synthetic intermediates. They are known to participate in carbonyl reductions, olefination reactions, organometallic additions, nucleophilic substitutions, and other reactions, allowing the preparation of various organofluorine skeletons without a loss of enantiopurity. ?,?,?
Sulfoxonium ylides are a class of compounds characterized by a carbanion directly attached to a positively charged sulfoxonium moiety. ?−? ? ? Due to this unique structure, they can act as both a nucleophile (through carbanion attack) and an electrophile (with neutral DMSO acting as a leaving group) within the same reaction. ?−? ? ? This dual reactivity makes them prime candidates for α,α-difunctionalization reactions with halogens. The first studies exploring this reactivity pattern date back to 1964, ?,? but a more comprehensive methodology has only been developed recently. In 2017 and 2021, our research group published consecutive studies detailing α,α-difunctionalization reactions with sulfoxonium ylides, leading to the preparation of a variety of highly functionalized products. ?,? This strategy eliminates ambiguity, allowing the use of one nucleophilic halogen and one electrophilic halogen within the same reaction vessel. However, despite these advances, all examples have been limited to the production of racemic products. As part of our group’s ongoing efforts to explore sulfoxonium ylide chemistry in asymmetric transformations, ?−? ? we describe herein the first asymmetric one-step α,α-chlorofluorination of α-carbonyl sulfoxonium ylides by two different modes: (a) the use of chiral Johnson‘s amino-sulfoxonium ylides 1 and (b) the use of Shibata’s chiral fluorinating agents, based on cinchona alkaloids (Schemeb). It is worth noting that in the case of Johnson’s amino-sulfoxonium ylides, this is the first demonstration of these important ylides being able to produce products with high enantiomeric excesses. ?,?
We started our work employing the (+)-(R)-(dimethylamino)ethyl-p-toIyloxosulfonium tetrafluoroborate salt, previously described by Johnson (see the Supporting Information for its preparation). ?,? Acylation with benzoyl chloride in the presence of NaH provided ylide 1 in 94% yield and 98:2 enantiomeric ratio. The absolute configuration of ylide 1h was determined by single-crystal X-ray diffraction (SCXRD) analysis (Figure S1). Chiral ylide 1a was then subjected to the chlorofluorination reaction under several reaction conditions and different sources of electrophilic/nucleophilic halogens to provide product 3a (Table).
We began our optimization studies using MeCN as the solvent for the reaction at room temperature and 5 °C, with entry 2 providing a superior result. Next, different solvents (entries 3–5) and KCl as a chloride source (entries 6 and 7) were screened, but no enhancement of the enantioselectivity was observed. However, the source of the electrophilic fluoride proved to have a profound impact on the enantioselectivity. First, using NFSI instead of Selectfluor (entry 8) did not result in improvement, but with pyridine-based “F^+^” reagent F3, the product was formed with 75% yield and 95:5 er (entry 9). Using this reagent at lower and higher temperatures (entries 10–12) and in combination with different solvents (entries 13–16) did not provide better selectivity of the product compared to entry 9 but did afford a reduced yield. With an efficient condition for the asymmetric chlorofluorination reaction (95:5 er) from chiral ylide 1a, we next extended our investigation by preparing 14 new ylides (1a–1n). These ylides were prepared in high enantiomeric ratios and then subjected to the conditions depicted in entry 9 of Table (Scheme).
2-Furyl product 3b was obtained with moderate enantioselectivity (86:14 er). para-substituted amino-sulfoxonium ylides furnished products 3c–3g and 3k with similar and >86:14 enantiomeric ratios, with the strong electron-withdrawing nitro group resulting in the lowest enantioselectivity (3d) and the unsubstituted benzene ring resulting in the highest observed enantioselectivity (3a, 95:5 er). These results imply that the electronics of the aromatic ring do not have a pronounced effect on the enantioselectivity of the reaction. The o-Cl-substituted ylide resulted in similar enantioselectivity, albeit in lower yield (3h, 19% yield, 85:15 er). Naphthyl-substituted ylide furnished product 3i in 91% yield and 91:9 er. Styryl-substituted product 3j was obtained in good yield but lower enantioselectivity (79%, 70:30 er). This result indicates that the steric hindrance or the π-system of the aromatic ring plays an important role in the interaction between the substrate and fluorinating agent in the enantio-determining step. Using ylides with α-propyl substitutions, products 3l and 3m were obtained with enantioselectivities of up to 92:8 er. To evaluate the effect of substituents on the sulfur chiral center of the ylide, we synthesized ylide 1n, which contains ethyl substituents on the nitrogen atom of the sulfoximine. When ylide 1n was subjected to the optimal conditions for α,α-dihalogenation, compound 3a was obtained in a reduced yield (60% vs 75%) and with a slightly lower er (94:6). These results indicate that a bulkier substituent on the nitrogen atom of the sulfoximine does not strongly influence the reaction selectivity. Exchanging F3 with NBS, product 4a was obtained in 51% yield and 96:4 er, showing that α,α-bromochlorination is also possible using this method. This result is particularly interesting, since it opens up the possibility of employing other combinations of electrophiles and nucleophiles in this reaction, possibly not limited to halogens. Although this reaction system is not catalytic, it has an advantage compared with classic chiral auxiliary reactions, since there is no need for an extra removal step. The absolute configuration of compounds 3 could not be determined since none of the synthesized compounds were solid and a crystal structure could not be obtained. Nevertheless, during our attempts at α,α-bromochlorination, we synthesized compound 4b in 70% yield, which was isolated as a solid. The enantiomeric ratio of this compound could not be determined, as we were unable to separate the two enantiomers. A crystal of this compound was obtained, and single-crystal X-ray diffraction (SCXRD) analysis was performed. The crystal structure of one of the enantiomers of 4b is displayed on Scheme and in detail in Figures S2 and S3.
Next, we were able to perform an asymmetric first-step formation of a C–F bond using an in situ preformed cinchona alkaloid fluorinating agent (Shibata’s classic procedure). ?−? ? The results obtained with this strategy are displayed in Table S4 (see discussion and the table in the Supporting Information). Several alkaloids in combination with the fluorine source were studied, with (QD)2_PHAL Q3 and Selectfluor providing the best results (61%, 89:11 er; acetone at −23 °C). With these best conditions in hand, we began to evaluate the behavior of substituted ylides under the reaction conditions (Scheme). Strong and mild electron-releasing groups -OMe and -Me in the para position furnished α,α-chlorofluoro products in good yields and good to moderate enantioselectivities (3c and 3n, respectively). On the other hand, strong electron-withdrawing group p-NO_2 resulted in lower yield and enantioselectivity (3d, 30%, 73:27 er). Halogen substitution on the aromatic ring furnished products with 84:16 and 81:19 er (3e and 3f, respectively). The 4-phenyl-substituted ring provided good enantioselection and yield (3g, 65%, 90:10 er). The 2-furyl derivative and ortho-substituted ylides were not good substrates for this reaction, delivering products in lower selectivities (3h and 3b, respectively). The naphthyl substituent provided product 3i in 51% yield and 79:21 er. To evaluate the robustness of our procedure, we performed the reaction on a 1.5 mmol scale. The reaction proceeded smoothly, providing product 3a in 65% yield (181.1 mg), albeit with a slightly lower enantioselectivity (85:15 er vs 89:11 er). Q3 was also recovered in 82% yield for the scaled-up reaction. We also evaluated a more diverse array of ylide structures, such as cyclic and acyclic esters and aryl–aryl-substituted keto ylide. We began evaluating aryl-ester ylides and found that for this class of substrates chiral amine C1 furnished better enantioselectivity than Q3. Phenyl ester derivative 3′a was prepared with 61:39 er in 48% yield. Other substituents on the ester moiety did not provide an improvement in enantioselectivity (61:39 er for 3′b, 56:44 er for 3′c). Cyclic ester ylide resulted in product 3′d with 70:30 er. Aryl–aryl-substituted ylides resulted in products with selectivities in the same range (69:31 er for 3′e, 71:29 er for 3′f). Mild electron-releasing and strong electron-withdrawing substituents at the para position of the aromatic ring for phenyl esters did not drastically alter the enantioselectivity (67:33 er for 3′g, 75:25 er for 3′h). Lastly, the reaction for the formation of product 3′i, bearing an acidic α-carbonyl hydrogen, resulted in a complex mixture of products. We also investigated the catalytic version from ylide 2a, employing 10–20 mol % catalyst. However, the enantiomeric ratios were reduced as well as the yields.
Lastly, additional tests regarding the interaction of the chiral aminosulfoxonium ylides with Q3 were performed. The use of ylides 1a and Q3, using Shibata’s chiral fluorinating agents, led to 3a in 86% yield and 85:15 er. Nonetheless, the use of ent-1a led to ent-3a in 89% yield and 78:22 er, a significant decrease in enantioselectivity compared to the other conditions (SchemeA). Although the tests did not led to an improvement in enantioselectivity, they showcase the divergence in interactions of the chiral sulfoxonium ylides with Q3. Treatment of 3a with an excess of Cl^–^ was performed to investigate if any stereoinversion would occur. After the elapsed time, no racemization was observed, indicating the formation of a stable stereocenter (SchemeB). It is worth noting that we evaluated several catalytic systems, including chiral organometallic complexes, chiral hydrogen bond donors, and chiral phase-transfer catalysts, for the α,α-dihalogenation of sulfur ylides. However, no enantioselectivity was observed, highlighting the challenging nature of this transformation. Further experiments can be performed to identify a suitable catalytic system for this reaction (see Tables S2 and S3 for full optimization conditions).
In summary, the first asymmetric one-step α,α-chlorofluorination of carbonyl compounds is presented. Combining two different strategies, 23 examples of α,α-chlorofluoro ketones or esters were prepared in an enantioenriched fashion. Two examples of α,α-bromochloroketones were also shown. Using the chiral amino-sulfoxonium strategy, 13 examples of α,α-chlorofluoro ketones were prepared with 19–90% yields and up to 95:5 er. Using the chiral fluorinating agent strategy, 19 examples of α,α-chlorofluoro carbonyl compounds were prepared in 19–80% yields and enantioselectivities of up to 90:10 er. Although the transformation is not catalytic, chiral promoter Q3 could be recovered in 82% yield and reused without a loss of enantioselection in the next reaction. Moreover, several different skeletons were evaluated, providing a more diverse study of α,α-chlorofluorination of carbonyl compounds. Comparing the products that were obtained by both strategies, it is clear that chiral aminosulfoxonium ylides proved to be superior, providing products in higher enantiomeric ratios (and similar or higher yields in every case). Furthermore, this is the first example in which Johnson’s aminosulfoxonium ylides have been used to obtain products with high enantiomeric excesses, underscoring the limited studies on this important class of sulfur ylides. We believe that this strategy can serve as a starting point for the development of other asymmetric difunctionalizations of aminosulfoxonium ylides using different combinations of electrophilic and nucleophilic heteroatoms, not limited to halogens. We believe that for both strategies, the mechanism is similar to that proposed for the racemic reaction,? with the initial attack of the sulfoxonium ylide on the electrophilic fluorine source generating the chiral carbon, which is then attacked by the chloride, with displacement of DMSO via an S_N_2 reaction. This hypothesis would explain the dramatic effect of the structure of the “F^+^” source on the observed er.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hagmann W. K.The Many Roles for Fluorine in Medicinal Chemistry J. Med. Chem.200851154359436910.1021/jm 800219 f 18570365 · doi ↗ · pubmed ↗
- 2Shah P.Westwell A. D.The Role of Fluorine in Medicinal Chemistry: Review Article Journal of Enzyme Inhibition and Medicinal Chemistry 200722552754010.1080/1475636070142501418035820 · doi ↗ · pubmed ↗
- 3Gillis E. P.Eastman K. J.Hill M. D.Donnelly D. J.Meanwell N. A.Applications of Fluorine in Medicinal Chemistry J. Med. Chem.201558218315835910.1021/acs.jmedchem.5b 0025826200936 · doi ↗ · pubmed ↗
- 4Inoue M.Sumii Y.Shibata N.Contribution of Organofluorine Compounds to Pharmaceuticals ACS Omega 2020519106331064010.1021/acsomega.0c 0083032455181 PMC 7240833 · doi ↗ · pubmed ↗
- 5Jeschke P.The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection Chem Bio Chem.20045557058910.1002/cbic.20030083315122630 · doi ↗ · pubmed ↗
- 6O’Hagan D.Harper B.D. Fluorine-Containing Natural Products J. Fluorine Chem.19991001–212713310.1016/S 0022-1139(99)00201-8 · doi ↗
- 7Wilkinson J. A.Recent Advances in the Selective Formation of the Carbon-Fluorine Bond Chem. Rev.199292450551910.1021/cr 00012 a 002 · doi ↗
- 8Champagne P. A.Desroches J.Hamel J.-D.Vandamme M.Paquin J.-F.Monofluorination of Organic Compounds: 10 Years of Innovation Chem. Rev.2015115179073917410.1021/cr 500706 a 25854146 · doi ↗ · pubmed ↗