Divergent Access to α,β-Unsaturated Thiolated and Selenolated Lactams via a Unified Palladium-Catalyzed Carbonylative Transformation
Zhiping Yin, Shuhui Sun, Xiaowen Qin, Heng Li, Fengxiang Zhu, Xiao-Feng Wu

TL;DR
A new method uses palladium to create lactam compounds with sulfur or selenium groups in one step.
Contribution
A unified palladium-catalyzed method for synthesizing thiolated and selenolated lactams from propargylamines.
Findings
Lactam core and sulfur/selenium groups are formed simultaneously under CO atmosphere.
Moderate to good yields achieved with functional group tolerance.
Propargylamine substrates with disulfide or diselenide reagents were successfully used.
Abstract
Herein, a unified and divergent palladium-catalyzed carbonylative procedure for the direct synthesis of α,β-unsaturated thiolated and selenolated lactams is reported. This strategy enables the simultaneous construction of the lactam core and installation of valuable sulfur or selenium functionalities via a single catalytic operation. By employing simple propargylamine substrates together with diphenyl disulfide or diselenide under a carbon monoxide atmosphere, the corresponding lactams were formed in moderate to good yields with promising functional group tolerance.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
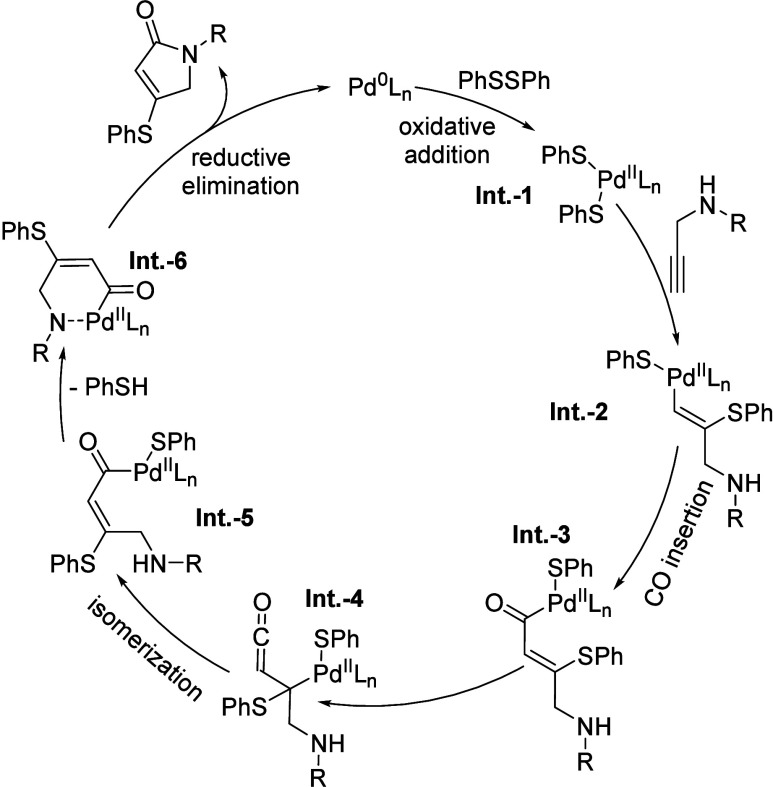 Figure 1
Figure 1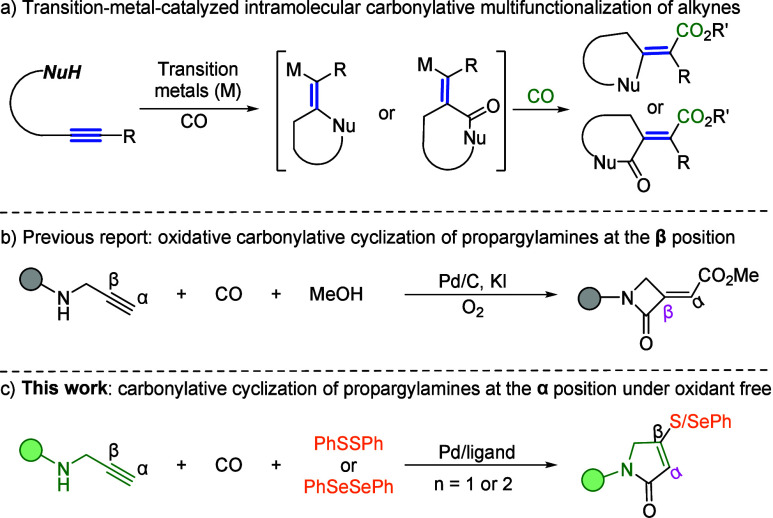 Figure 2
Figure 2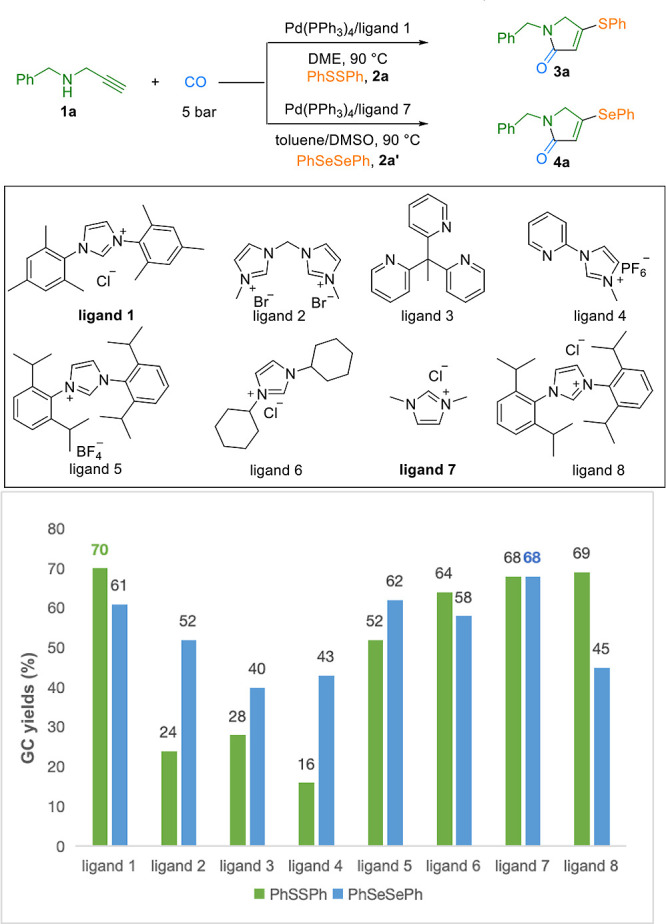 Figure 3
Figure 3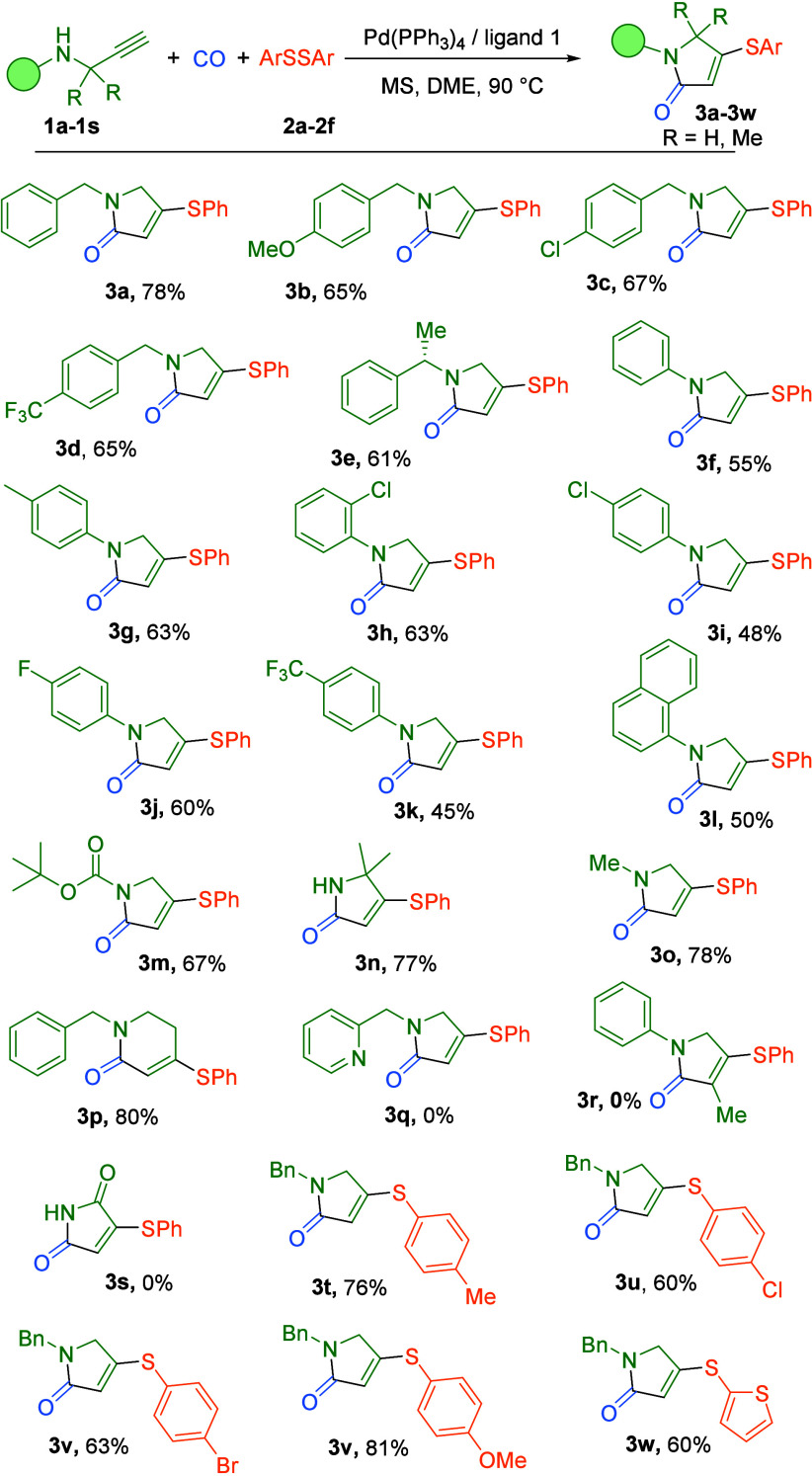 Figure 4
Figure 4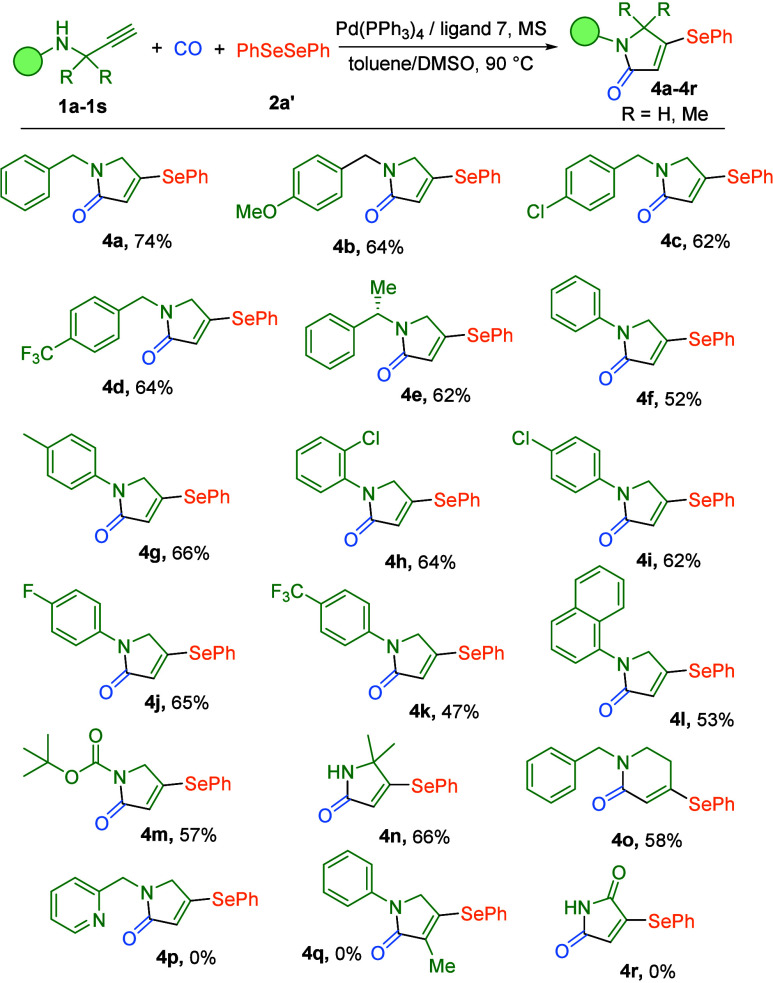 Figure 5
Figure 5 Figure 6
Figure 6 Figure 7
Figure 7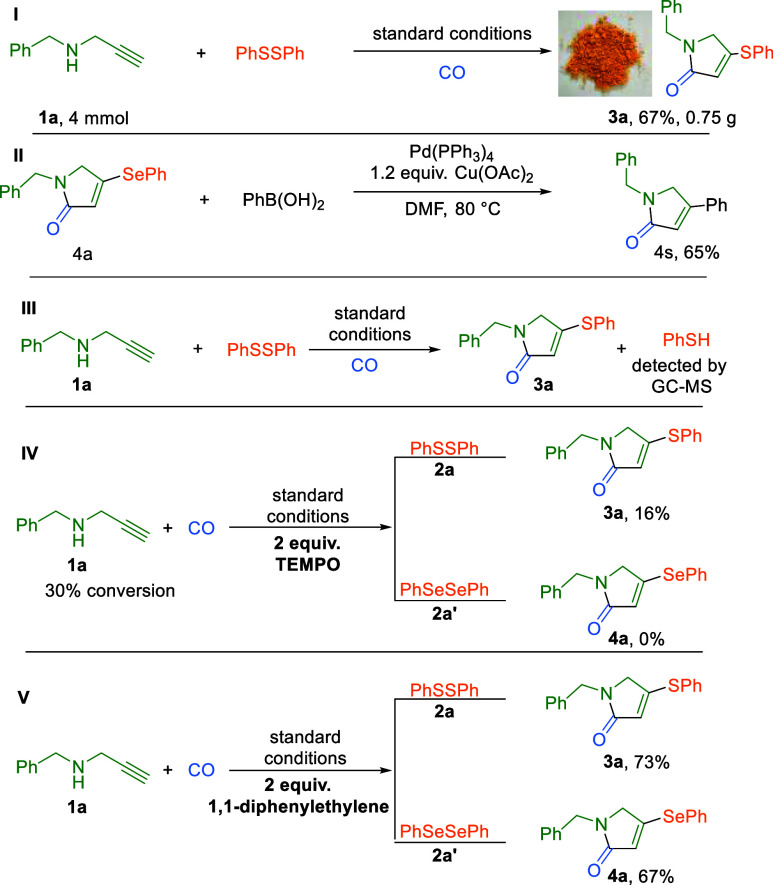 Figure 8
Figure 8- —Senior Talent Foundation of Jiangsu University10.13039/501100004300
- —Natural Science Foundation of Jiangsu Province10.13039/501100004608
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSulfur-Based Synthesis Techniques · Organoselenium and organotellurium chemistry · Catalytic C–H Functionalization Methods
α,β-Unsaturated amides, particularly in the form of lactams, constitute a privileged structural motif in medicinal chemistry and chemical biology.? Their electrophilic α,β-unsaturated carbonyl system serves as a key handle for irreversible engagement with biological nucleophiles, such as cysteine residues, via conjugate addition, making them invaluable in the design of covalent inhibitors and bioactive probes.? Consequently, the development of efficient and modular synthetic methods to access diversely functionalized unsaturated lactams remains a high-priority objective in synthetic organic chemistry.
Transition-metal-catalyzed carbonylative cyclizations have emerged as a powerful and direct strategy for constructing heterocyclic cores from unsaturated precursors and CO gas (Schemea).? Among these transformations, the hydroformylative cyclization of alkyne-tethered amines represents a well-established route to lactams.? However, this approach is inherently constrained by its reliance on the incorporation of a hydrogen atom across the alkyne, thereby failing to introduce versatile functional handles that would enable further molecular diversification. In parallel, transition-metal-catalyzed intramolecular oxidative carbonylative difunctionalization of alkynes offers a complementary route to various heterocycles. A representative example is the work by Gabriele and co-workers, who achieved the synthesis of (Z)-α-(methoxycarbonyl)methylene-β-lactams from propargylamines using a catalytic system comprising PdI_2_ and KI (Schemeb).? While such oxidative carbonylative methodologies expand structural diversity, they predominantly follow a β-selective cyclization pathway and typically depend on external oxidants. Consequently, the direct and selective installation of valuable heteroatomic functionalitiessuch as sulfur or selenium, each of which is known to modulate pharmacokinetic profiles and serve as synthetic handles for further elaborationspecifically at the α-position of the alkyne during lactam formation, remains a significant and unmet challenge.?
Although notable progress has been made in intermolecular seleno- and thiocarbonylation of alkynes, demonstrating the utility of (PhSe)2 or (PhS)2 for assembling acyclic β-functionalized acrylates,? the translation of this difunctionalization logic into the more demanding context of intramolecular carbonylative cyclization has not been realized, representing a conspicuous gap in synthetic methodology. Notably, in 1997, Sonoda, Ogawa, and co-workers developed a palladium-catalyzed carbonylative cyclization of propargyl alcohols with diaryl diselenides and diaryl disulfide.? Various lactones were formed in moderate to good yields with Pd(PPh_3_)4 as the catalyst.
Inspired by these precedents and driven by our group’s long-standing focus on carbonylative heterocycle synthesis,? we now report a distinct catalytic paradigm that directly addresses this formidable challenge (Schemec). This method enables an oxidant-free, α-selective carbonylative cyclization of propargylamines, directly constructing the α,β-unsaturated lactam scaffold. The key innovation lies in the synergistic interception of the pivotal vinyl-palladium intermediate by dichalcogenides (PhSSPh or PhSeSePh). This seamless merger of cyclization with C–S or C–Se bond formation achieves a dual breakthrough. It fundamentally shifts the selectivity from the traditional β-position to the novel α-position of alkynes and concurrently installs valuable sulfur or selenium functionalities at the β-position of alkynes. This mild and modular protocol not only provides a unified route to previously elusive, densely functionalized heterocyclic building blocks but also exhibits remarkable divergence, enabling the programmable synthesis of both five- and six-membered lactams from a common set of starting materials simply by varying the tether length.
Building upon the established importance of selenolated lactams, we initiated our study by exploring the carbonylative cyclization of N-benzylprop-2-yn-1-amine (1a) with diphenyl diselenide (2a′) as a model substrate. While our initial attempt using molybdenum hexacarbonyl as a solid CO surrogate proved to be unsuccessful, switching to carbon monoxide gas enabled the formation of the desired 1-benzyl-4-(phenylselanyl)-1,5-dihydro-2H-pyrrol-2-one (4a). This preliminary conversion was achieved using Pd(PPh_3_)4 with additional PPh_3_ as a ligand in toluene at 90 °C (see the Supporting Information). Systematic screening of phosphine ligands following this initial result, however, did not lead to positive improvements in yield. A pivotal advancement came with the observation that N-heterocyclic carbene (NHC) ligands consistently outperformed their phosphine counterparts, which might be due to NHC coordinating more strongly with Pd. ?,? Guided by this finding, we evaluated several catalysts and additives (Table, entries 2–4, and Supporting Information). Although common additives showed limited effects, the introduction of molecular sieves (MS) notably enhanced the reaction efficiency. The possible reason might be the presence of moisture in the system, which led noncarbonylation or hydrosulfuration as the main side reaction. Capitalizing on the superior performance of NHC ligands, we subsequently screened a series of such ligands (Table) and identified ligand 7 as being optimal. Further optimization of the reaction medium led to the adoption of a toluene/DMSO mixed solvent system, ultimately delivering model product 4a in 74% yield. Here, we believe that the addition of DMSO can increase the solubility of the selenium reagent. Notably, due to the high coordinating and toxicity properties of sulfur to palladium compared with Se, more steric ligand is needed to achieve better results. With the optimized protocol for selenium incorporation established, we next examined the generality of our catalytic system by extending it to the analogous sulfurative cyclization using diphenyl disulfide (2a). Employing substrate 1a under comparable conditions, optimization of the ligand and solvent identified DME as the preferred solvent, with Pd(PPh_3_)4 and ligand 1 serving as cocatalysts under 5 bar of CO at 90 °C as the optimal setup for the thiolated lactam synthesis. The reaction efficiency for both transformations decreased dramatically in the absence of a catalyst, a ligand, or molecular sieves (Table, entries 8–10).
Under the optimized reaction conditions, we systematically evaluated the substrate scope of this palladium-catalyzed carbonylative cascade process. As summarized in Table, the transformation demonstrated excellent functional group compatibility, delivering a diverse array of α,β-unsaturated thiolated lactams in moderate to excellent yields.
Systematic evaluation of electronic effects revealed that benzylic alkynes bearing either electron-donating (MeO) or electron-withdrawing (Cl and CF_3_) substituents participated effectively in the transformation, affording the corresponding products in 65–67% yields (Table, 3b–3d). Notably, the reaction proved to be compatible with a chiral substrate derived from (S)-1-phenylethan-1-amine, affording the desired lactam in 61% yield (Table, 3e). The protocol was further extended to aromatic amine-derived alkynes bearing various substituents, with both electron-donating (Me) and electron-withdrawing groups (Cl, F, and CF_3_) being well tolerated (45–63% yields) (Table, 3f–3l). Investigation of amine protecting groups demonstrated that diverse nitrogen functionalities, including Boc-protected, N-methyl, and free amine variants, all underwent smooth conversion to the target lactams (67–78% yields) (Table, 3m–3o). Particularly significant was the successful formation of a six-membered lactam ring from a homoallylic amine substrate in 80% yield, highlighting the method’s versatility in ring-size control (Table, 3p). For pyridine-substituted alkyne substrates, internal alkynes, and amide-linked alkynes, the carbonylative transformation proved to be ineffective, yielding no target product (Table, 3q–3 s). To further establish the reaction generality, we examined the scope of disulfide coupling partners using alkyne 1a as the standard substrate. The method accommodated various para-substituted diphenyl disulfides (Me, Cl, Br, and OMe) with comparable efficiency (60–81% yields) (Table, 3t–3v). Importantly, heteroaromatic 1,2-di(thiophen-2-yl)disulfane also participated effectively, delivering the thiolated product in 60% yield (Table, 3w).
Subsequently, recognizing the significant biological profiles of selenolated lactams, we extended our catalytic system to the direct construction of this valuable scaffold. As summarized in Table, this selenium variant exhibited excellent compatibility with the full range of alkyne substrates previously investigated. Benzyl alkynes bearing electronically diverse substituents (MeO, Cl, and CF_3_) delivered the corresponding selenolated products in 62–74% yields, showing the same efficiency compared to their sulfur analogues (Table, 4a–4d). The chiral (S)-1-phenylethan-1-amine-derived alkyne also provides the selenolactam in 62% yield (Table, 4e). Aromatic amine-tethered alkynes with both electron-donating and -withdrawing groups afforded products in 47–66% yields, demonstrating consistent electronic tolerance (Table, 4f–4l). Various nitrogen protecting groups (Boc, free NH_2_) all proved to be viable under the selenium conditions, affording products in 57–66% yields (Table, 4m and 4n). Notably, the six-membered ring formation proceeded with acceptable efficiency, achieving 58% yield from the homoallylic amine substrate (Table, 4o). This systematic evaluation establishes the robust generality of our catalytic system for selenium incorporation.
To demonstrate the practical utility of this methodology, a gram-scale reaction was performed at a 4 mmol scale, affording thiolated lactam 3a in 67% isolated yield with efficiency comparable to that of the small-scale standard reaction (Scheme, I). This result confirms the robustness and potential scalability of the protocol for industrial applications. Furthermore, the synthetic versatility of the selenolated lactam products was explored through a palladium-catalyzed Suzuki–Miyaura cross-coupling with phenylboronic acid, constructing a new C–C bond in 65% yield (Scheme, II). These successful transformations highlight the value of the α,β-unsaturated lactams as versatile synthetic intermediates and further establish the broad utility of the present carbonylative cascade strategy.
To gain deeper mechanistic insight into the carbonylative transformation, we performed a series of control experiments (Scheme, III–V). Under the standard conditions, the key intermediate benzenethiol, derived from S–S bond cleavage of 2a, was successfully detected by GC–MS analysis (Scheme, III). The reaction pathway was further probed using radical scavengers, including TEMPO and 1,1-diphenylethylene (Scheme, IV). In the reactions with TEMPO, the yield of the desired products was inhibited significantly. However, due to the oxidizing property of TEMPO, we could not fully confirm the presence of the radical intermediate. Notably, the addition of 1,1-diphenylethylene did not suppress the transformation, affording products 3a and 4a in 73% and 67% yields, respectively, thereby ruling out the involvement of radical intermediates (Scheme, V).
Based on these experimental results and relevant literature precedents, ?,? a plausible catalytic cycle is proposed in Figure. The cycle is initiated by oxidative addition of the S–S bond in 2a to in situ-generated Pd(0), furnishing Pd(II) thiolate complex Int-1. Subsequent coordination and regioselective insertion of the terminal alkyne into the Pd–S bond afford vinyl-palladium species Int-2. Carbon monoxide coordination and insertion then yield acyl-palladium intermediate Int-3, which undergoes stereoselective isomerization to give (E)-Int-5. Finally, the intramolecular cyclization of (E)-Int-5 releases the lactam product along with benzenethiol, regenerating the active Pd(0) catalyst to close the cycle.
In conclusion, we have developed a unified Pd-catalyzed carbonylative procedure that divergently delivers α,β-unsaturated thiolated or selenolated lactams from readily available propargylamines and dichalcogenides. This transformation establishes a distinct α-selective cyclization mode, contrasting with conventional β-selective carbonylative cyclization pathways, and concurrently introduces synthetically versatile PhS or PhSe groups at the alkenyl position. The method exhibits a broad substrate scope and scalable performance and enables further structural diversification of the lactam products through cross-coupling transformation. Beyond providing direct access to previously challenging heterocyclic architectures, this work introduces a programmable and multifunctionalization-enabled approach to carbonylative cyclization, opening new avenues for the efficient assembly of bioactive molecule scaffolds.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Zhang S. K.Neumann H.Beller M.Synthesis of α,β-unsaturated carbonyl compounds by carbonylation reactions Chem. Soc. Rev.2020493187321010.1039/C 9CS 00615 J 32255444 · doi ↗ · pubmed ↗
- 2a Ramachandran P. V.Yip-Schneider M.Schmidt C. M.Natural and synthetic α,β-unsaturated carbonyls for NF-κB inhibition Future Medicinal Chemistry 2009117920010.4155/fmc.09.1521426075 · doi ↗ · pubmed ↗
- 3a Perrone S.Troisi L.Salomone A.Heterocycle Synthesis through Pd-Catalyzed Carbonylative Coupling Eur. J. Org. Chem.201920194626464310.1002/ejoc.201900439 · doi ↗
- 4a Lightburn T. E.De Paolis O. A.Cheng K. H.Tan K. L.Regioselective Hydroformylation of Allylic Alcohols Org. Lett.2011132686268910.1021/ol 200782 d 21504208 PMC 3096926 · doi ↗ · pubmed ↗
- 5Bonardi A.Costa M.Gabriele B.Salerno G.Chiusoli G. P.Versatile synthesis of beta-lactams, gamma-lactams or oxalines by palladium-catalysed oxidative carbonylation of 1-substituted prop-2-ynylamines Tetrahedron Lett.1995367495749810.1016/0040-4039(95)01514-0 · doi ↗
- 6a Gallo-Rodriguez C.Rodriguez J. B.Organoselenium Compounds in Medicinal Chemistry Chem Med Chem 202419 e 20240006310.1002/cmdc.20240006338778500 · doi ↗ · pubmed ↗
- 7a Zhu F.Wu H.Wu X.-F.Palladium/rhodium-catalyzed four-component carbonylative difunctionalization of alkynes: Regio- and stereoselective esterification/selenylation to access β-selenyl acrylates J. Catal.202544811617010.1016/j.jcat.2025.116170 · doi ↗
- 8a Yin Z.Xu J.Wu X.-F.No Making Without Breaking: Nitrogen-Centered Carbonylation Reactions ACS Catal.2020106510653110.1021/acscatal.0c 01479 · doi ↗