Severe COVID‐19 Unveils Atypical Familial Hemophagocytic Lymphohistiocytosis due to a Novel Homozygous PRF1 Variant
Nasimeh Vatandoost, Mohsen Jari, Mansour Salehi

TL;DR
A rare PRF1 gene variant was found in a child with severe complications from COVID-19 and other infections, revealing a genetic condition linked to family history.
Contribution
A novel homozygous PRF1 variant is identified as a cause of atypical familial hemophagocytic lymphohistiocytosis triggered by SARS-CoV-2.
Findings
A novel PRF1 variant was identified through whole-exome sequencing in a child with severe disease.
The PRF1 variant was inherited from asymptomatic parents and predicted to be harmful.
Severe complications and death occurred despite treatment, highlighting the role of viral triggers in revealing genetic conditions.
Abstract
We report a novel homozygous PRF1 variant, PRF1 (NM_001083116.3):c.343G > A (p.Glu115Lys), identified by whole‐exome sequencing (WES) in an 11‐year‐old girl with atypical familial hemophagocytic lymphohistiocytosis (FHL). The variant, inherited from asymptomatic heterozygous parents, was absent or extremely rare in population databases and was predicted to be deleterious by multiple in silico tools. Born to consanguineous parents, the patient presented with recurrent fever, pancytopenia, and multiorgan failure following SARS‐CoV‐2 infection, further complicated by Epstein–Barr virus (EBV) and cytomegalovirus (CMV) coinfections. Despite intensive immunosuppressive therapy, she developed seizures, an intracranial hemorrhage, and died at age 11. A striking family history of unexplained febrile deaths in infancy and childhood strongly supported autosomal recessive inheritance. It emphasizes…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
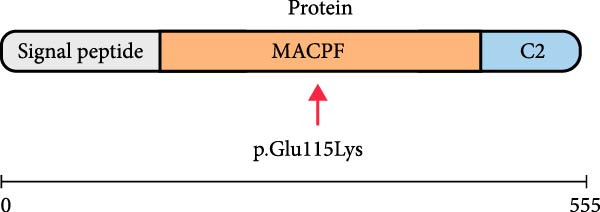 Figure 1
Figure 1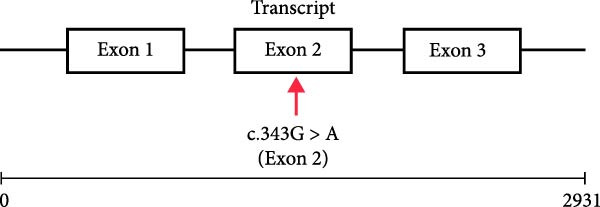 Figure 2
Figure 2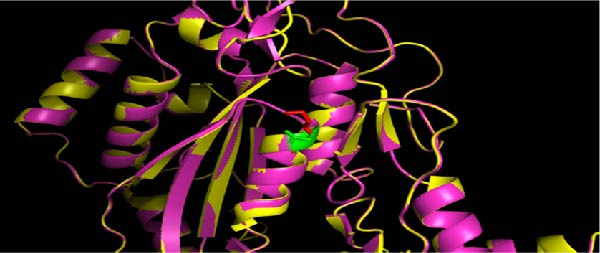 Figure 3
Figure 3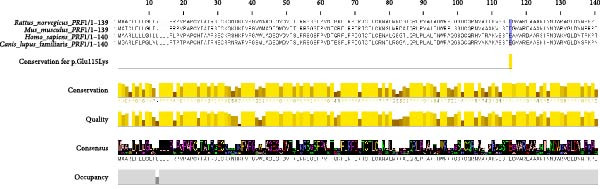 Figure 4
Figure 4 Figure 5
Figure 5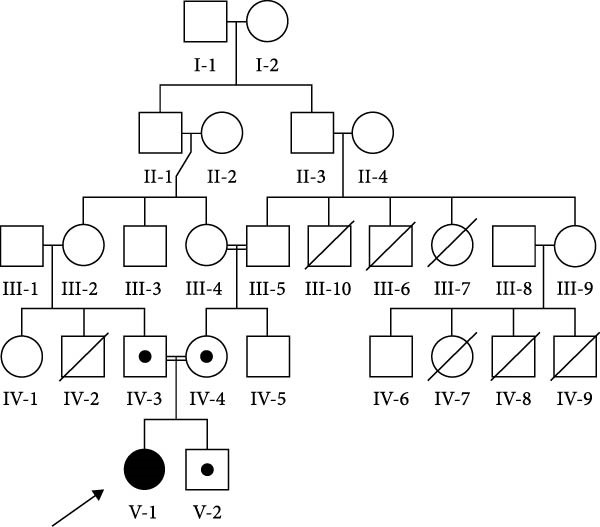 Figure 6
Figure 6| Primer name | Sequence (5′–3′) | Product size (bp) |
|---|---|---|
|
| ATTCCAGAGCCCAAGTGC | — |
|
| GCTGAAGCTGTACTGGTCC | 540 |
| Parameter | Initial lab results | Monitoring during periods of disease inactivity | Fever + pancytopenia (admission) | Discharge | Significant fever episode | Reference range |
|---|---|---|---|---|---|---|
| Hemoglobin (× 109/L) | 1.58 (L) | 7.95 | 0.81 (L) | 2.56 (L) | 1.80 (L) | 108–133 |
| Platelet (× 109/L) | 33 (L) | 136 (L) | 30 (L) | 46 (L) | 24 (L) | 194–345 |
|
WBC (× 109/L) | — | — | — | — | — | 4.5–13.5 |
| Neutrophile (× 109/L) | 0.80 (L) | 3.98 | 0.26 (L) | 1.80 (L) | 1.09 (L) | 1.82–7.47 |
| Lymphocytes (× 109/L) | 0.90 (L) | 2.54 | 0.35 (L) | 0.49 (L) | 0.37 (L) | 1.16–3.33 |
| Monocytes (× 109/L) | 0.46 | 0.80 (H) | 0.11 (L) | 0.24 (L) | 0.08 (L) | 0.19–0.72 |
| Eosinophil (× 109/L) | 0.05 | 0.24 | 0.03 (L) | 0.01 (L) | 0.00 (L) | 0.02–0.32 |
| Basophilia (× 109/L) | 0.00 (L) | 0.00 (L) | 0.00 (L) | 0.01 | 0.00 (L) | 0.01–0.05 |
| Alanine transaminase(U/L) | 28 | ‐ | 171 (H) | ‐ | 127 (H) | < 31 |
| Aspartate aminotransferase (U/L) | 17 | ‐ | 395 (H) | ‐ | 331 (H) | < 31 |
| Fibrinogen(g/L) | — | 2.6 | 2.0 | — | 1.5 (L) | 1.9–4.3 |
| Ferritin (µg/L) | 351.4 (H) | 239.6 (H) | 638.8 (H) | 687.6 (H) | 4873.6 (H) | 13.7– 78.8 |
| Triglycerides (mmol/L) | ‐ | 2.38 (H) | 2.99 (H) | 2.06 (H) | 5.49 (H | < 1.02 |
| CRP (mg/L) | ‐ | ‐ | ‐ | 108.8 | 200 | < 10 |
| E.S.R 1 h | 15 mm/h | — | — | — | — | — |
| CMV qualitative PCR | ‐ | ‐ | ‐ | ‐ | Positive‐116.9 copies/mL | Negative |
| EBV qualitative PCR | ‐ | ‐ | ‐ | ‐ | Positive‐3328 copies/mL | Negative |
| Tool/database | Score | Classification (threshold) | References |
|---|---|---|---|
| SIFT | 0.02 | Damaging ( ≤ 0.05) | [ |
| PolyPhen‐2 | 0.98 | Probably damaging ( ≥ 0.85) | [ |
| MutationTaster | 1.0 | Disease‐causing (1.0) | [ |
| CADD (Phred) | 17.09 | Deleterious ( ≥ 10) | [ |
| FATHMM‐MKL | 0.9223 | Damaging ( ≥ 0.5) | [ |
| BayesDel (addAF) | 0.1077 | Pathogenic ( ≥ 0.057) | [ |
| gnomAD v2.1.1 1000Genomes, GME, and Iranome (allele frequency) | < 0.0001 | Rare (supportive of PM2) | [ |
| Stability prediction server | INPS‐3D | mCSM | DUET | DDGun | SDM | SAAFEC‐SEQ | I‐Mutant2.0 |
|---|---|---|---|---|---|---|---|
|
| Destabilizing | Destabilizing | Destabilizing | Destabilizing | Destabilizing | Destabilizing | Destabilizing |
| Gene | ClinVar status | Inheritance | Zygosity (sibling) | Zygosity (parents) | Zygosity (proband) | Variant (protein) | Variant (cDNA) | ACMG classification |
|---|---|---|---|---|---|---|---|---|
|
| Uncertain significant (SCV002175656.3, SCV005152109.1) | Autosomal recessive | Heterozygous | Heterozygous | Homozygous | p.Glu115Lys | Likely pathogenic |
- —Isfahan University of Medical Sciences10.13039/501100003970
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research · Immunodeficiency and Autoimmune Disorders · Neurogenetic and Muscular Disorders Research
1. Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life‐threatening hyperinflammatory syndrome caused by uncontrolled immune activation [1]. Primary familial HLH (FHL) typically presents in infancy due to biallelic pathogenic variants in genes encoding cytotoxic proteins, including PRF1, UNC13D, STX11, and STXBP2 [2–5]. However, late‐onset and adult presentations are increasingly recognized [6–8]. The PRF1 gene encodes perforin, essential for pore formation and target‐cell killing by NK cells and cytotoxic T lymphocytes. Loss‐of‐function variants in PRF1 (FHL2) account for 20%–50% of FHL cases [9]. Recently, SARS‐CoV‐2, the virus responsible for COVID‐19, has emerged as a potent trigger for HLH. It exacerbates underlying genetic vulnerabilities, thereby complicating diagnosis and treatment. The atypical presentations pose significant diagnostic and therapeutic challenges because of shared symptoms with other autoimmune and hyperinflammatory disorders [10–12].
We report an 11‐year‐old patient whose condition worsened after COVID‐19 and showed signs of HLH. Whole‐exome sequencing (WES) identified a novel homozygous PRF1 mutation, which was subsequently confirmed by Sanger sequencing and cosegregation analysis. This study highlights the importance of genetic testing in atypical HLH cases and emphasizes the role of viral triggers, like SARS‐CoV‐2, in revealing genetic predispositions to hyperinflammatory conditions. Our findings expand the known PRF1 mutations associated with HLH and underscore the need for personalized diagnosis and treatment in such complex cases.
2. Materials and Methods
2.1. Case Presentation and DNA Extraction
The patient’s medical history, clinical data, and family pedigree were documented following genetic counseling. Peripheral blood was collected with informed consent from the legal guardian, and the study was approved by the Ethics Committee of Isfahan University of Medical Sciences (IR.MUI.MED.REC.1401.006). Genomic DNA was extracted using the FAVORGEN Blood Genomic DNA Extraction Mini‐Kit (Tehran, Iran).
2.2. WES and Variant Prioritization
WES was performed on the proband’s DNA using the Illumina NovaSeq x 10B platform (2 × 150 bp, 300‐cycle configuration) with the xGen Exome Research Panel v2 (Integrated DNA Technologies). Sequencing achieved a mean depth of 100x, with over 90% of target bases at 20x coverage and over 95% at a Q30 quality score. Reads were aligned to GRCh38 using BWA‐MEM, and variants were called with GATK HaplotypeCaller. Variants were prioritized and interpreted in accordance with the ACMG/AMP guidelines [13].
2.3. In Silico and Structural Analysis
Variant pathogenicity was evaluated using SIFT, PolyPhen‐2, MutationTaster, CADD, FATHMM‐MKL, and BayesDel, with allele frequencies checked in gnomAD v2.1.1, Iranome, and GTEx databases. Conservation analysis of the p.(Glu115Lys) variant within the MACPF domain was performed using Clustal Omega v1.2.4 (https://www.ebi.ac.uk/tools/msa/clustalo/) with default settings (including multiple sequence alignment iterations and four iterations), and the results were visualized using Jalview v2.11.1.5. Protein stability was assessed with INPS‐3D, mCSM, DUET, DDGun, SDM, SAAFEC‐SEQ, and I‐Mutant2.0. Homology models were built using SWISS‐MODEL (https://swissmodel.expasy.org) with template PDB 3NSJ and visualized in PyMOL v3.1.6.1 (https://pymol.org).
2.4. Sanger Sequencing and Cosegregation Analysis
Sanger sequencing confirmed the candidate mutation, and cosegregation analysis was performed on the family. Primers (PRF1‐F/R) were designed to amplify a 540‐bp region containing the variant and were validated using online tools, including Primer‐BLAST and SNP Check (Gene Tools, SNP Check V3). The primer sequences used are listed in Table 1.
3. Result
3.1. Subject
An 11‐year‐old girl, born to consanguineous parents, had an unremarkable prenatal and postnatal history with normal development until age 6. She experienced minor, unexplained fevers that resolved on their own until she developed abdominal pain and fever at age 6, initially attributed to stress. After contracting COVID‐19 at age 6, she presented with high‐grade fever, pancytopenia, and splenomegaly confirmed by ultrasound. Laboratory results showed elevated liver enzymes (ALT:171 U/L and AST:395 U/L), borderline thrombocytopenia (30 × 10^9^/L), increased triglycerides (2.99 mmol/L), and high ferritin levels (638.8 µg/L) (Table 2). She was hospitalized repeatedly every 1–2 months with symptoms of fever, joint pain, and cytopenia, fulfilling part of the HLH diagnostic criteria [14]. She was initially started on intravenous immunoglobulin (IVIG) for its anti‐inflammatory effects due to an uncertain diagnosis and concerns about the potential for worsening infections. Additionally, high‐dose intravenous glucocorticoids were initiated. A few months later, the patient began to report progressive muscular weakness in her lower limbs, leading to frequent falls. During her hospitalization, she developed seizures, and a repeat head CT scan showed a new intracranial hemorrhage with herniation. At this point, she was treated with high doses of intravenous glucocorticoids, cyclosporin A, and dexamethasone. Throughout her hospitalization, she also required supportive therapy, including leukocyte‐depleted red blood cells, platelet units, and frozen plasma. Serological testing confirmed Epstein–Barr virus (EBV; IgM‐positive, 3328 DNA copies/mL) and cytomegalovirus (CMV;116.9 copies/mL) infections, with retinal inflammation causing severe vision loss. Despite 4 years of treatment, the patient died at age 11, just before planned intrathecal chemotherapy. The family medical history was notable for the deaths of five cousins who suffered from undiagnosed illnesses characterized by prolonged fever. All of these cousins died during their first or second year of life.
Additionally, the mother’s uncles also passed away due to a fever complicated by jaundice, with ages at death ranging from 1 to 20 years. The father’s brother also died because of repetitive, unexplained seizures. Unfortunately, details regarding the treatment regimens and investigations of deceased patients are unavailable (Figure 1A).
Figure 1(A) Pedigree showing autosomal recessive inheritance. The proband (II‐1, arrow) is homozygous (filled symbol) with atypical HLH and a fatal outcome; parents (I‐1 and I‐2) and sibling (II‐2) are heterozygous (half‐shaded); five cousins, two maternal uncles, and one paternal uncle (shaded with diagonal lines) had unexplained febrile deaths or seizures. (B) Sanger sequencing electropherograms confirming the PRF1 (NM_001083116.3):c.343G > A (p.Glu115Lys) variant: homozygous in the proband, heterozygous in parents and healthy sibling, supporting cosegregation (PP1and ACMG).(A)(B)
3.2. Genetic Findings
WES identified a homozygous variant PRF1 (NM_001083116.3):c.343G > A (p.Glu115Lys). Sanger sequencing confirmed homozygosity in the proband and heterozygosity in both parents and the healthy sibling (Figure 1B). The variant is absent from gnomAD v4 and Iranome. All in silico tools predicted pathogenicity (Table 3). Conservation analysis identified Glu115 as conserved across species (Figure 2A). Predicted protein structural changes based on SWISS‐MODEL homology models of wild type and mutant Perforin‐1 yielded GMQE = 0.92 and QMEAN = −0.85. PyMOL v2.5 alignment yielded an RMSD of 0.054 Å across 516 of 555 residues. Visualization of NP_001076585.2:p.(Glu115Lys) in the MACPF domain’s β‐sheet, a charge reversal from glutamate to lysine, was displayed (Figure 2B). Stability predictions indicated a destabilizing effect (Table 4).
Figure 2. Conservation and structural analysis of the PRF1 p.(Glu115Lys) variant. (A) Conservation analysis of Glu115 across species, performed using clustal omega and visualized with Jalview, showing high conservation in the MACPF domain. (B) Structural visualization of the NP_001076585.2:p.(Glu115Lys) variant in the MACPF domain using PyMOL. Cartoon and surface views highlight the charge shift from glutamate (negative, green) to lysine (positive, red) in the β‐sheet, with an RMSD of 0.054 Å for aligned residues.(A)(B)
3.3. ACMG Classification
The variant PRF1 (NM_001083116.3):c.343G > A p.(Glu115Lys) is located in Exon 2 of the transcript and within the MACPF domain of the PRF1 protein (Figure 3). It was classified as a variant of uncertain significance (VUS) in ClinVar by two laboratories (Invitae SCV002175656.3 and Ambry SCV005152109.1) based solely on its rarity and in silico predictions. We contacted both laboratories on September 18, 2025, to share phenotypic data; Invitae declined due to privacy restrictions, and Ambry did not respond. According to the full ACMG/AMP guidelines, it meets the following criteria: PM2 (allele frequency less than 0.0001 in gnomAD, 1000 Genomes, Greater Middle East [GME], and Iranome, indicating the rarity of the variant, as shown in Table 3), PM1 (located in the conserved MACPF domain, Figure 3), PP1 (cosegregation with HLH in the family, heterozygous in a healthy sibling and unaffected parents), PP3 (multiple in silico tools predict damaging effects, Table 3), and PP4 (phenotype specific for HLH).
Figure 3(A) Transcript NM_001083116.3 showing exons and c.343G > A in exon 2. (B) Protein NP_001076585.2 domains with p.(Glu115Lys) in the MACPF domain.(A)(B)
4. Discussion
This case describes an 11‐year‐old female with atypical HLH triggered by SARS‐CoV‐2, linked to a novel homozygous variant PRF1 (NM_001083116.3):c.343G > A (p.Glu115Lys). The patient’s late‐onset presentation, with recurrent fevers, pancytopenia, and multiorgan dysfunction postCOVID‐19, aligns with reports of FHL manifesting beyond infancy [3, 5]. The three extensive family histories of unexplained febrile deaths and seizures (Figure 1A) support a genetic etiology, consistent with autosomal recessive inheritance confirmed by cosegregation analysis (Figure 1B).
The PRF1 (NM_001083116.3):c.343G > A (p.Glu115Lys) variant, located in the conserved MACPF domain (Figure 2A), was predicted to be pathogenic by multiple in silico tools (Table 3), with an allele frequency of less than 0.0001 in gnomAD, supporting its rarity (PM2 and ACMG). Structural analysis with SWISS‐MODEL and PyMOL (RMSD = 0.054 Å, 516 out of 555 residues aligned) showed minimal overall structural deviation but a significant change in charge at residue 115, from glutamate (negative) to lysine (positive), within the MACPF domain’s β‐sheet (Figure 2B). This charge reversal, along with stability predictions indicating a destabilizing effect (Table 4), suggests a potential disruption of electrostatic interactions or hydrogen bonds, which are essential for perforin’s pore‐forming function. Such disruptions may impair NK cell and CTL cytotoxicity, contributing to the hyperinflammation observed in HLH [25].
The patient’s clinical decline after COVID‐19, worsened by EBV and CMV coinfections, underscores the influence of viral triggers in uncovering genetic predispositions in FHL [10, 11, 26, 27]. SARS‐CoV‐2 hyperinflammatory effects, which resemble cytokine release syndrome, likely worsened the underlying perforin dysfunction, resulting in severe HLH manifestations [12].
The family history of early deaths (Figure 1A) aligns with patterns in FHL pedigrees, where consanguinity increases homozygous variant prevalence [12]. In our case, cosegregation analysis, where the homozygous mutation was inherited from both asymptomatic parents, aligns with autosomal recessive inheritance patterns observed in other familial HLH cases, further supporting the pathogenicity of the identified variant.
Despite intensive immunosuppressive therapy (IVIG, glucocorticoids, cyclosporine A, and dexamethasone), the patient’s fatal outcome underscores the importance of early genetic diagnosis and hematopoietic stem cell transplantation (HSCT), which is the only curative treatment for FHL2 [28, 29]. Early HSCT greatly enhances survival, especially when done before severe organ damage [30].
The ACMG classification of likely pathogenic (Table 5) is supported by rarity (PM2), conservation (Figures 2 and 3; PM1), cosegregation (PP1), in silico predictions (PP3), and HLH‐specific phenotype (PP4). However, the absence of functional assays (PS3 criterion) limits definitive causality, as in silico and structural analyses alone cannot confirm perforin’s functional impairment [13]. Future studies should incorporate NK cell cytotoxicity or perforin expression assays to validate the variant’s impact, potentially guiding the timing of HSCT [31]. This case emphasizes the diagnostic challenges of atypical HLH in older children and the critical role of genetic screening in identifying novel PRF1 mutations. The interplay between genetic predisposition and viral triggers, particularly SARS‐CoV‐2, underscores the need for integrated genomic and immunological approaches to enhance outcomes in FHL.
5. Limitations
The absence of functional assays to evaluate the impact of the p. (Glu115Lys) variants on perforin activity precludes application of the ACMG PS3 criterion. In silico predictions (Table 3), structural analysis (Figure 2B), and cosegregation data (Figure 1B) support pathogenicity, but further assays, such as NK cell cytotoxicity or protein expression assays, are required to establish causality.
6. Conclusion
Severe SARS‐CoV‐2 infection unmasked late‐onset FHL2 in an 11‐year‐old girl carrying the novel homozygous variant PRF1 (NM_001083116.3):c.343G > A (p.Glu115Lys), here classified as likely pathogenic despite current ClinVar VUS status. This case underscores that primary HLH must remain in the differential diagnosis of older children with virus‐triggered hyperinflammation, particularly in consanguineous families. Prompt genetic testing for PRF1 and related genes is essential, as HSCT offers the only curative option.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported by a grant from the Isfahan University of Medical Sciences (Grant IR.MUI.MED.REC.1401.006).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sham M. , Zhu R. , and Pasternak Y. , Whole Exome Sequencing Identifies Causative Compound Heterozygous Variants in PRF 1 in Late-Onset Familial Hemophagocytic Lymphohistiocytosis, Lympho Sign Journal. (2022) 9, no. 4, 85–91.
- 2Meeths M. , Horne A. C. , Sabel M. , Bryceson Y. T. , and Henter J.-I. , Incidence and Clinical Presentation of Primary Hemophagocytic Lymphohistiocytosis in Sweden, Pediatric Blood & Cancer. (2015) 62, no. 2, 346–352, 10.1002/pbc.25308, 2-s 2.0-84927509752.25382070 · doi ↗ · pubmed ↗
- 3Sienes Bailo P. , Goñi Ros N. , and Menéndez Jándula B. , et al.First Case of Very Late-Onset FHL 2 in Spain With Two Variants in the PRF 1 Gene, Annals of Clinical Biochemistry. (2023) 60, no. 5, 356–364, 10.1177/00045632231186076.37365821 · doi ↗ · pubmed ↗
- 4Janka G. E. , Familial and Acquired Hemophagocytic Lymphohistiocytosis, European Journal of Pediatrics. (2007) 166, no. 2, 95–109, 10.1007/s 00431-006-0258-1, 2-s 2.0-33845878531.17151879 · doi ↗ · pubmed ↗
- 5Stadermann A. , Haar M. , and Riecke A. , et al.Late Onset of Primary Hemophagocytic Lymphohistiocytosis (HLH) With a Novel Constellation of Compound Heterozygosity Involving Two Missense Variants in the PRF 1 Gene, International Journal of Molecular Sciences. (2024) 25, no. 5, 10.3390/ijms 25052762, 2762.38474010 PMC 10931657 · doi ↗ · pubmed ↗
- 6Côte M. , Ménager M. M. , and Burgess A. , et al.Munc 18-2 Deficiency Causes Familial Hemophagocytic Lymphohistiocytosis Type 5 and Impairs Cytotoxic Granule Exocytosis in Patient NK Cells, The Journal of Clinical Investigation. (2009) 119, no. 12, 3765–3773, 10.1172/JCI 40732, 2-s 2.0-72849125357.19884660 PMC 2786810 · doi ↗ · pubmed ↗
- 7Gholam C. , Grigoriadou S. , Gilmour K. C. , and Gaspar H. B. , Familial Haemophagocytic Lymphohistiocytosis: Advances in the Genetic Basis, Diagnosis and Management, Clinical and Experimental Immunology. (2011) 163, no. 3, 271–283, 10.1111/j.1365-2249.2010.04302.x, 2-s 2.0-79551703613.21303357 PMC 3048610 · doi ↗ · pubmed ↗
- 8Zur Stadt U. , Rohr J. , and Seifert W. , et al.Familial Hemophagocytic Lymphohistiocytosis Type 5 (FHL-5) is Caused by Mutations in Munc 18-2 and Impaired Binding to Syntaxin 11, American Journal of Human Genetics. (2009) 85, no. 4, 482–492, 10.1016/j.ajhg.2009.09.005, 2-s 2.0-70350500464.19804848 PMC 2756548 · doi ↗ · pubmed ↗