A Medically Managed Case of Acromegaly: A Case Report
Karam Bdour, Khaldon Al-Sarihin, Nesreen El Issa, Odai Alwraikat, Mohammad Albadarneh, Rania Al-Asa'd, Mu'taz Alwadi

TL;DR
This case report describes a 39-year-old man with acromegaly successfully treated with medical therapy, showing improvement in hormone levels and tumor size.
Contribution
The novelty lies in demonstrating the effectiveness of combined medical therapy for a large pituitary tumor without surgery.
Findings
The patient showed significant biochemical and anatomical improvement with medical therapy.
Combined use of somatostatin analogs and dopamine agonists was effective in managing the tumor.
Abstract
Endocrine glands are specialized cells responsible for producing and secreting hormones that serve many functions in various body tissues. The pituitary gland is an endocrine gland located in the brain, composed of anterior and posterior parts, both of which are responsible for the secretion of many hormones. The anterior pituitary gland secretes six hormones. Hypersecretion and undersecretion result in abnormalities in the body. Autonomous growth hormone production due to a pituitary adenoma leads to various changes in tissues and organs. Excess growth hormone results in gigantism during childhood, while in adulthood, it is called acromegaly. In this report, we present a 39-year-old man with a large somatotroph macroadenoma, which was managed entirely with medical therapy, resulting in significant biochemical and anatomical improvement. This case highlights the potential role of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
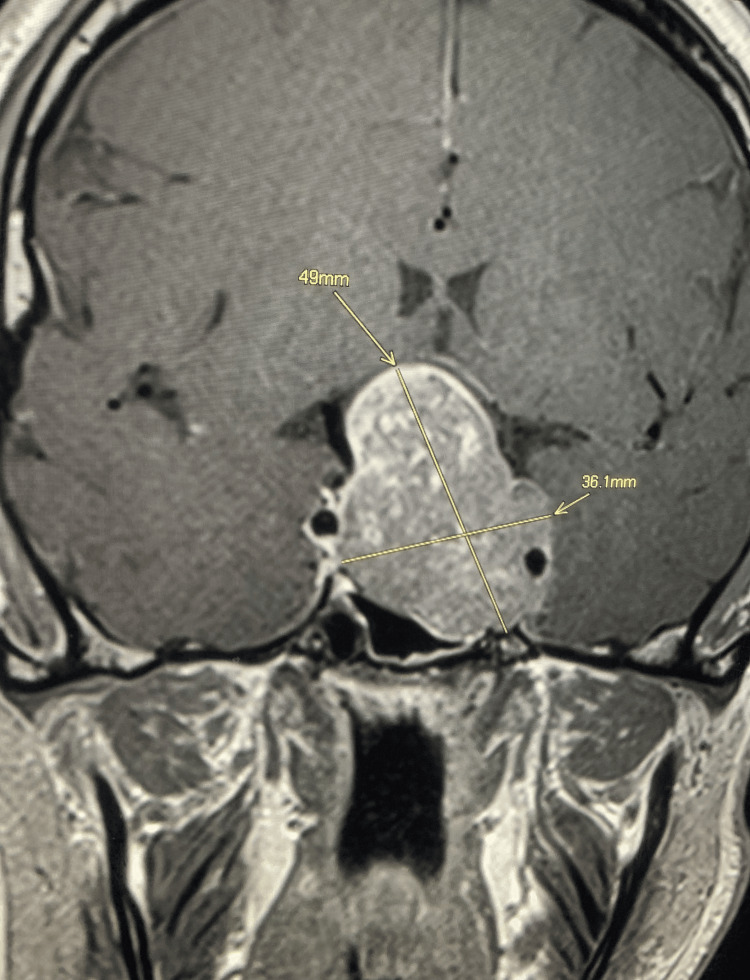 Figure 1
Figure 1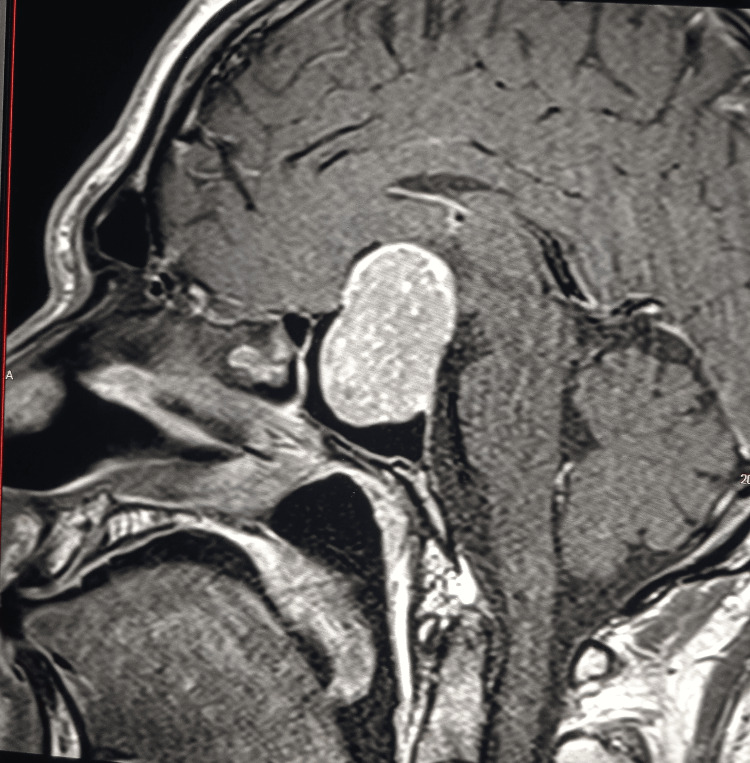 Figure 2
Figure 2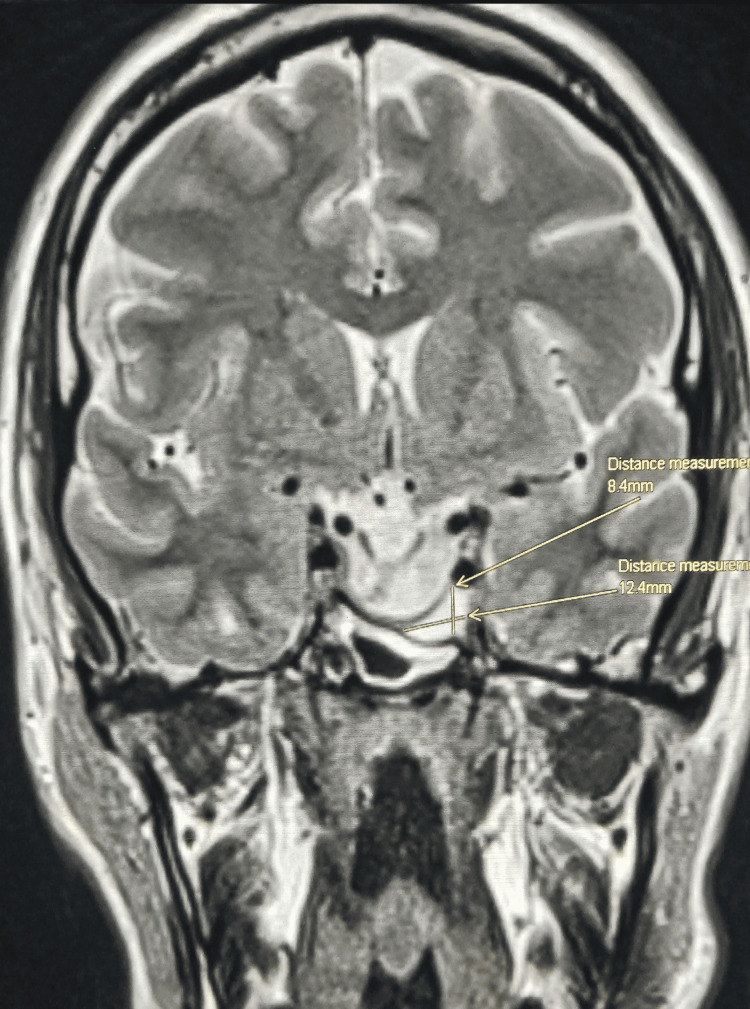 Figure 3
Figure 3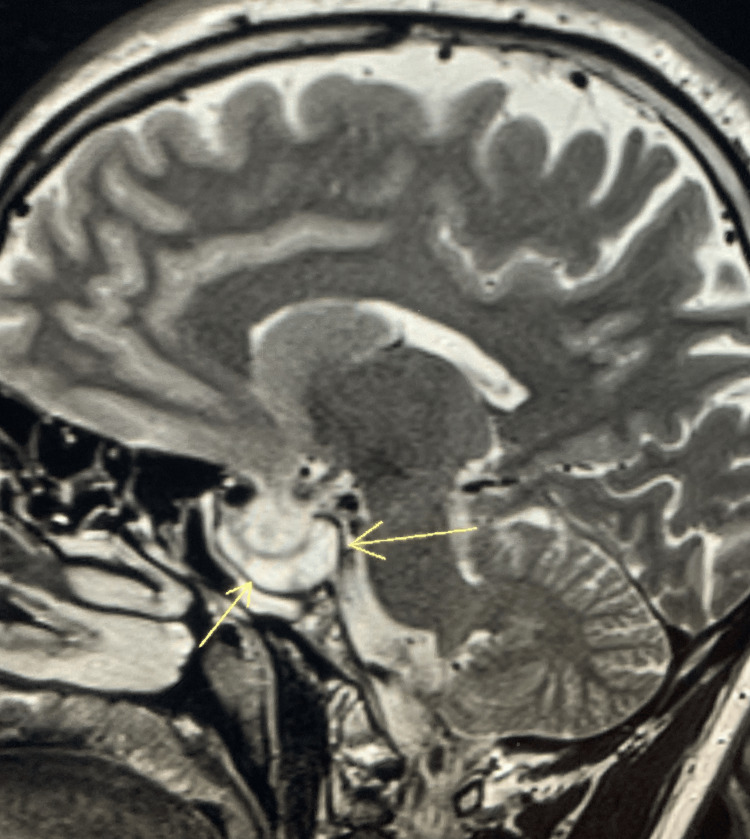 Figure 4
Figure 4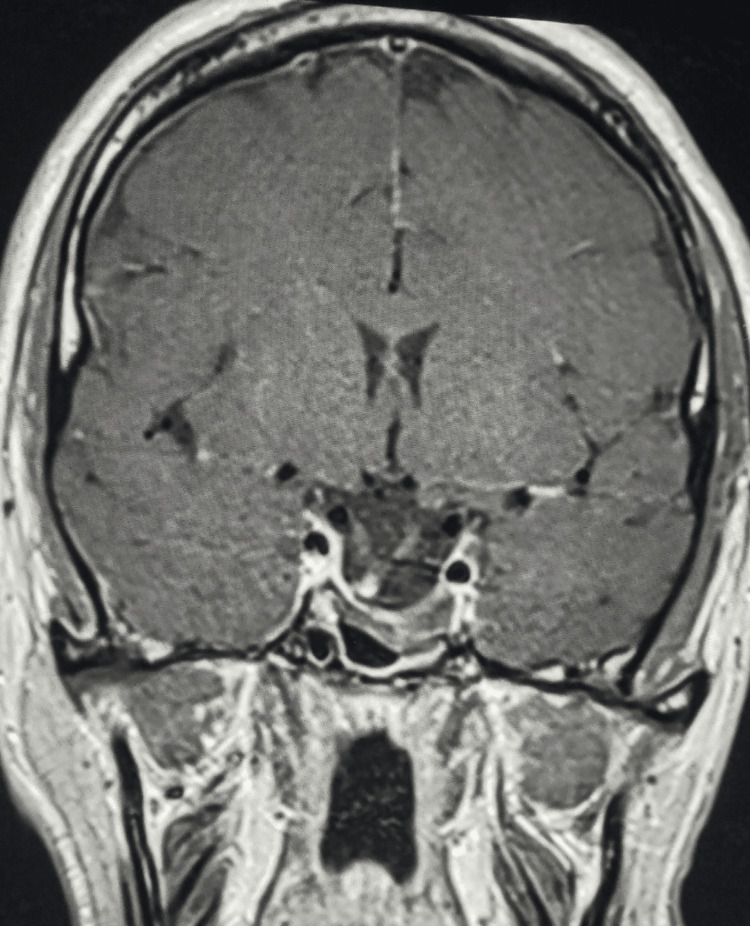 Figure 5
Figure 5| 75 g oral glucose tolerance test (min) | Baseline | 30 | 60 | 90 | 120 |
| Growth hormone (ng/mL) | 21.5 | 8.7 | 6.1 | 5.2 | 10.5 |
| Blood glucose (mg/mL) | 81 | 130 | 111 | 126 | 146 |
| Initial levels | Three months post-treatment | Six months post-treatment | 12 months post-treatment | Normal values | |
| GH | 22.7 | 15.7 | 13.9 | 4.5 | 0.06-5 ng/mL |
| IGF-1 | 784 | 567 | 343 | 256 | 220-307 ng/mL |
| Prolactin | 440 | 276 | 157 | 17.6 | 4-15 ng/mL |
| TSH | 1.33 | 1.59 | 1.4 | 1.07 | 0.27-4.2 µIU/mL |
| T4 | 0.6 | 1.3 | 1.4 | 1.2 | 0.7-1.7 ng/dL |
| ACTH | 57 | 38.8 | 38 | 47.6 | 7.2-63 pg/mL |
| Cortisol | 8.25 | 12.6 | 11.67 | 13.8 | 7.0-25 ug/dL |
| LH | 2.1 | 2.7 | 3.6 | 3.5 | 1.3-9.6 mIU/mL |
| FSH | 1.8 | 1.5 | 1.9 | 3.1 | 1.2-15.8 mIU/mL |
| Total testosterone | 73.6 | 49 | 201 | 278 | 246-836 ng/dL |
| HbA1c | 6.4% | 6.1% | 5.8% | 5.7% | Normal < 5.6% |
| FBS | 111 | 105 | 106 | 98 | 90-100 mg/dL |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPituitary Gland Disorders and Treatments · Adrenal and Paraganglionic Tumors · Adrenal Hormones and Disorders
Introduction
The anterior part of the pituitary gland is responsible for the formation and release of six hormones, each of which has a specific function. The blood levels of these hormones are orchestrated based on positive and negative feedback [1,2]. The uncontrollable release of growth hormone (GH) results in gigantism or acromegaly. Acromegaly is a disease characterized by many clinical manifestations that occur in adulthood (after bony epiphyseal plates fusion) due to excessive GH secretion [1,3,4].
Acromegaly has a prevalence of between 2.8 and 13.7 cases per 100,000 population, and it has no sex difference; the average age of diagnosis is around forty [5,6].
The most common cause of this disease is a pituitary adenoma with excess secretion of GH. Rare cases of GH excess can be observed in neuroendocrine tumors, which produce GH-releasing hormone (GHRH), stimulating uncontrollable GH release from the pituitary gland [7].
Diagnosing acromegaly is suspected when there is a high clinical suspicion based on many features, such as an increase in the size of body tissues.
Acromegaly should be diagnosed biochemically and structurally by a high insulin-like growth factor 1 (IGF1) level, along with clinical features, and further workup, including an oral glucose tolerance test (OGTT) for GH and the presence of a pituitary adenoma on pituitary imaging using contrast-enhanced magnetic resonance imaging [7].
As GH and prolactin both originate from somatotroph cells, some cases of GH excess have prolactin co-secretion. Around a quarter of patients diagnosed with acromegaly have prolactin hormone excess [8-10].
Once confirmed biochemically and structurally, the gold standard treatment is transsphenoidal adenectomy [3,7].
Case presentation
We represent Mr. X, a 39-year-old male who presented to the endocrine clinic at King Hussain Medical Center after an incidental diagnosis of pituitary adenoma during evaluation of chronic headaches. Consent was taken from the patient to publish his case, but he did not accept publishing personal images.
Upon evaluation, his blood pressure was 141/83 with a normal temperature and heart rate. He had coarse facial features, protrusion of the mandible, macroglossia, a mild, smooth, non-nodular goiter, and bulky hands. His visual fields were assessed using a confrontation test, which revealed the presence of bitemporal hemianopia. After taking a detailed history, he noted that he had changed his shoe size from 42 to 44 over the last three years; additionally, he reported experiencing a long-standing headache accompanied by diminished vision. When asked about his sexual life, he reported experiencing low libido and erectile dysfunction. The patient reported that his wedding ring no longer fit due to increased finger size.
His vision was assessed by visual perimetry, and bitemporal hemianopia was also shown.
A provisional diagnosis of acromegaly was suspected. Thus, a proper workup was initiated, including laboratory and radiological tests.
Laboratory and radiological diagnosis
The hormonal panel of the pituitary gland indicated elevated levels of GH and IGF-1. The IGF-1 level was 742 ng/mL, and the GH level was 22.7 ng/mL. Prolactin was 440 ng/mL, thyroid-stimulating hormone (TSH) was 2.1 µIU/mL, cortisol was 8.9 (µg/dL), and adrenocorticotropic hormone (ACTH) was 39 (pg/mL). Both luteinizing hormone (LH) and follicle-stimulating hormone (FSH) were at low normal levels (mIU/mL).
Free T4 (µg/mL) level was 0.6, and total testosterone level was around 180 µg/dL in two morning readings (normal range 246-836 ng/dL). Regarding blood sugar, his HbA1c was 6.4% and fasting blood sugar was 115 mg/dL.
According to the 2014 Endocrine Society Clinical Practice Guideline on acromegaly, the gold standard diagnostic test is the lack of suppression of GH to < 1 ng/mL after ingesting 75 g of the OGTT. Consequently, the patient underwent the OGTT, and it confirmed the diagnosis of acromegaly [3]. Table 1 shows the OGTT result.
A two-dimensional transthoracic echocardiography showed mild left ventricular hypertrophy with normal ejection fraction and no valvopathy.
Magnetic resonance imaging of the pituitary gland revealed a very large macroadenoma measuring 48 × 38 × 29 mm, with hemorrhagic components pushing the optic chiasm superiorly and extending both inferiorly and superiorly, while laterally invading the cavernous sinuses (Figures 1-2).
Pituitary MRI - coronal view Before the treatment, showing the initial dimensions of the lesion.
Pituitary MRI - sagittal viewBefore the treatment.
Treatment
Treatment lines were discussed with the patient. This includes initially surgical treatment as the gold standard treatment, medical treatment (somatostatin analogues, pegvisomant, and dopamine receptor agonists) as the second line, and radiotherapy as the third line [3,5].
As surgical management is the first line of treatment in such cases, the patient was given the first dose of somatostatin analogues, had the low free T4 level replaced with thyroxine 75 mcg/day, and was referred for neurosurgery for the surgical option.
The procedure, course, and outcomes were reviewed with the patient, and an experienced surgeon advised the transcranial approach owing to the large size of the macroadenoma and its extension into adjacent tissues. However, the patient declined surgery due to perceived risks and was hesitant to undergo surgery via the transcranial approach.
The patient was referred back to the endocrine clinic for a different management module. The medical treatment options in the patient's case were discussed, and the medical plan was 120 mg lanreotide long-acting release (LAR) monthly via deep subcutaneous injection [3,5]. Due to the presence of co-secretion, a dopamine agonist was added to his treatment plan [11]. Using cabergoline 2 mg per week. Because of the huge size of his macroadenoma, pegvisomant was avoided as it might cause tumor growth and put more pressure on adjacent tissues [12].
The patient was periodically assessed using imaging and a biochemical profile during his follow-up. His vision had improved more markedly peripherally, and some of his symptoms had disappeared. Such as the resolution of his headache, better sexual drive and activity, and he also observed that his hands were getting smaller. His blood pressure was around 110/70 during multiple clinic visits.
After 12 months of medical treatment, the size of his pituitary macroadenoma decreased by almost 75%, measuring 12 × 10 × 8 mm abutting the optic chiasm. The patient’s IGF1 dropped to 253, and prolactin to 10.8 ng/dL. Figures 3-5 demonstrate the shrinkage in size.
Pituitary MRI - coronal viewTwelve months after treatment, with the dimensions of lesion.
Pituitary MRI - sagittal viewTwelve months after treatment.
Pituitary MRI - coronal viewTwelve months after treatment.
The dramatic improvement in his symptoms and his IGF1/prolactin levels, along with structural regression of tumor size, was all achieved by medical treatment only [13-15].
There are some cases reported in the literature for successful shrinkage of somatotroph macroadenoma, but they needed more time [16-20]. Probably the addition of a dopamine agonist to the medical management helped to enhance the structural shrinkage of the somatotroph adenoma and shortened the duration needed for medical success in this case [11,21,22]. Table 2 shows the hormonal changes during the course of management.
Discussion
Acromegaly is an uncommon disorder with gradual manifestations; usually, the patient presents due to acromegaly's systemic complications in the body organs [3]. Acromegaly is a slow-developing condition that can go unnoticed for a long time, which often results in a late diagnosis or treatment.
Over the years, the sustained hypersecretion of GH has resulted in cardiovascular compromise, respiratory dysfunction, higher malignancy risk, metabolic disorders, skeletal deformities, arthropathy, and neuropathies. Therefore, it is not unusual for the consequences of acromegaly to increase mortality in affected individuals [7]. The cardiovascular changes (cardiomyopathy) account for the major causes of mortality [23]. That is why most acromegaly patients present to different medical specialties before the disease is diagnosed [24].
Acromegaly is mostly caused by pituitary adenoma [2-4,7]. Some rare causes result from ectopic secretion of GH or GHRH by different tumors [7,25]. Prolactin is co-secreted in approximately 25% of individuals with acromegaly [8-10].
When diagnosed, a GH-secreting pituitary adenoma is the main cause of acromegaly. Based on tumor size and the presence of optic chiasm compression, visual field disturbances may occur along with systemic manifestations of GH oversecretion [3,7].
Adenectomy, specifically via the transsphenoidal approach, appears to be the initial treatment for most patients. Suppose surgery is not considered as a primary treatment, such as in patients with advanced age, fragile medical status, or the patient’s preference [26]. Medical therapy, including somatostatin analogs, can result in adequate treatment by suppressing the autonomous secretion of GH with subsequent inhibition of IGF-1 production by the liver and controlling the effects of GH hypersecretion on various body tissues [13-15]. Pegvisomant is also used as part of the medical management of acromegaly; it blocks the receptor of GH, which, as a result, decreases the IGF-1 level. However, such a mechanism can increase the size of the pituitary adenoma and might put more compression on the surroundings of the pituitary gland [12]. Dopamine agonists can also be part of the medical treatment of acromegaly [11,21,22]. The use of dopamine agonists showed marked control in cases of acromegaly manifested by normalization of IGF-1 level and mass reduction of the pituitary adenoma, especially in prolactin co-secreting adenomas [22-29].
The term silent pituitary adenoma is not applicable in this case because the patient had overt acromegaly manifested clinically and biochemically, as the GH and IGF-1 were both elevated throughout the disease management [30].
Mr. X presented with overt acromegalic features, and after proper and targeted assessment, the acromegaly was confirmed biochemically. The MRI of the pituitary gland revealed a large pituitary adenoma compressing the adjacent structures.
In this case report, we highlight the outcome of medical treatment in an acromegaly patient, and how it resulted in structural effects on pituitary adenoma size, and biochemical normalization of IGF-1 level and prolactin level.
As the patient was reluctant to undergo adenectomy, the medical treatment was the next option according to the treatment guidelines [3,5]. The use of somatostatin analogues along with a dopamine agonist for 12 continuous months showed a significant decrease in somatotroph adenoma size, and the IGF1 concentration reached a normal level. The prolactin level also dropped to 15 ng/dL.
The patient's clinical features of acromegaly syndrome have almost resolved completely; however, he still has some facial coarsening. The patient had successful management using medical modalities only.
Limitations include the single-patient nature of the report and relatively short follow-up, but the findings add to growing evidence supporting medical therapy as a viable primary option in selected cases.
Conclusions
Acromegaly is frequently diagnosed late due to its slow progression and nonspecific manifestations. When a pituitary adenoma is the cause, transsphenoidal surgical resection remains the cornerstone of treatment, while medical therapy is generally reserved for specific circumstances, such as patient refusal or persistent disease after surgery.
This case demonstrates that in selected patients, an exclusive medical therapy with lanreotide and cabergoline can achieve excellent biochemical control and meaningful tumor shrinkage. Timely diagnosis, individualized treatment planning, and close follow-up are essential to optimize outcomes and reduce long-term complications of GH excess. Larger studies and longer follow-up are needed to confirm these findings, but this case adds to the growing evidence that individualized medical management can be both safe and effective in acromegaly.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Physiology, pituitary hormones Stat Pearls Sadiq NM Tadi P Treasure Island, FL Stat Pearls Publishing 2023 https://www.ncbi.nlm.nih.gov/books/NBK 55755632491488 · pubmed ↗
- 2Cellular and molecular specificity of pituitary gland physiology Physiol Rev Perez-Castro C Renner U Haedo MR Stalla GK Arzt E 138922012 https://doi.org/10.1152/physrev.00003.20112229865010.1152/physrev.00003.2011 · doi ↗ · pubmed ↗
- 3Acromegaly: an endocrine society clinical practice guideline J Clin Endocrinol Metab Katznelson L Laws ER Jr Melmed S Molitch ME Murad MH Utz A Wass JA 393339519920142535680810.1210/jc.2014-2700 · doi ↗ · pubmed ↗
- 4Gigantism and acromegaly Stat Pearls Bello MO Garla VV Treasure Island, FL Stat Pearls Publishing 2025 https://www.ncbi.nlm.nih.gov/books/NBK 538261/30855849 · pubmed ↗
- 5A comprehensive review of four clinical practice guidelines of acromegaly Cureus Ogedegbe OJ Cheema AY Khan MA 014202210.7759/cureus.28722 PMC 945386936105896 · doi ↗ · pubmed ↗
- 6Epidemiology of acromegaly: review of population studies Pituitary Lavrentaki A Paluzzi A Wass JA Karavitaki N 492020172774317410.1007/s 11102-016-0754-x PMC 5334410 · doi ↗ · pubmed ↗
- 7Acromegaly Endocrinol Metab Clin North Am Ben-Shlomo A Melmed S 1011223720081822673210.1016/j.ecl.2007.10.002PMC 2697616 · doi ↗ · pubmed ↗
- 8Hyperprolactinemia in acromegaly is related to prolactin secretion by somatolactotroph tumours Horm Metab Res Van Laethem D Michotte A Cools W Velkeniers B Unuane D Andreescu CE Bravenboer B 6476535220203275718710.1055/a-1207-1132 · doi ↗ · pubmed ↗