Therapeutic targeting of Aurora Kinase A in advanced prostate cancer
Maroun Bou Zerdan, Mehmet Asim Bilen, Jindan Yu

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
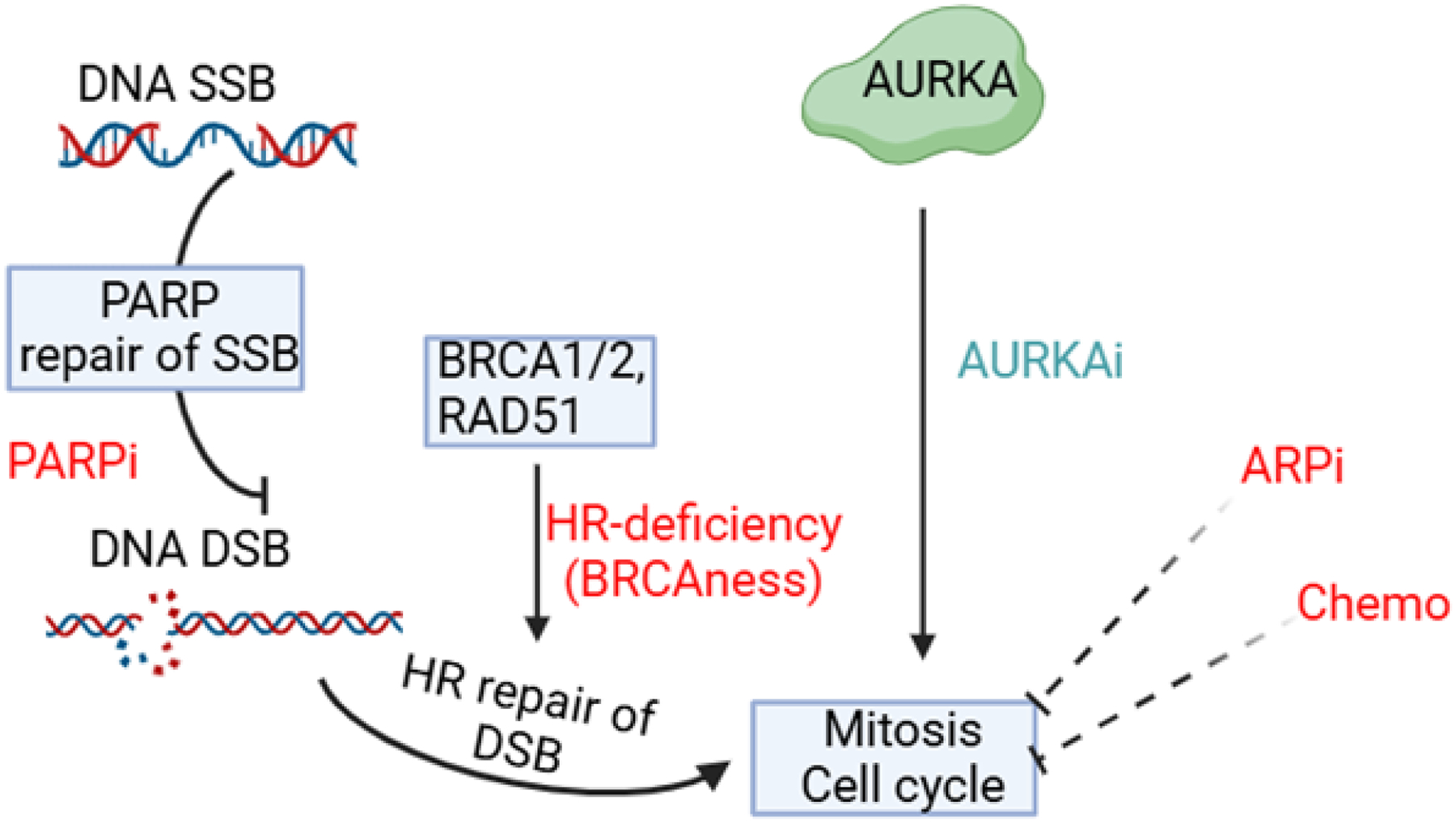 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrotubule and mitosis dynamics · Ubiquitin and proteasome pathways · Mechanisms of cancer metastasis
Commentary
Prostate cancer (PCa) typically progresses from an androgen-dependent state to a more aggressive form known as castration-resistant prostate cancer (CRPC) following androgen deprivation therapy (ADT). ADT, which includes surgical or medical castration, initially reduces tumor burden by inhibiting androgen receptor (AR) signaling, a critical driver of prostate cancer growth [1, 2]. However, CRPC eventually develops due to several mechanisms that restore or bypass AR signaling. These mechanisms include AR gene amplification, AR mutations, expression of constitutively active AR splice variants, and intratumoral androgen synthesis [1–3]. The molecular mechanisms underlying this transition involve genetic, epigenetic, and hormonal changes that promote cellular plasticity. Key genetic alterations include the loss of tumor suppressor genes such as RB1, TP53, and PTEN, and changes in epigenetic regulators like EZH2, which facilitate the lineage switch from prostate adenocarcinoma to NEPC [4, 5]. Additionally, transcription factors such as ASCL1 and SOX2 play crucial roles in driving neuroendocrine differentiation and maintaining the NE phenotype [6, 7]. The development of NEPC often follows extensive androgen receptor pathway inhibitors, antiandrogen resistance, and a loss of androgen receptor expression. Despite castrate levels of circulating androgens, these adaptations allow continued AR activity, driving tumor progression [1, 2]. A subset of CRPC tumors, however, can become independent of AR signaling and adopt neuroendocrine features, leading to neuroendocrine prostate cancer (NEPC). The emergence of NEPC is associated with resistance to conventional therapies and poor prognosis. NEPC is characterized by the loss of AR expression and the gain of neuroendocrine markers, such as chromogranin A and synaptophysin [4]. This transition is often driven by genetic and epigenetic changes, including the loss of tumor suppressors like TP53 and RB1, the activation of lineage plasticity pathways, with concomitant upregulation of cell cycle drivers including MYCN, PLK1, Cyclin D1, and Aurora kinase A (AURKA) [8–10].
AURKA is a serine/threonine kinase that controls the timing of mitotic entry and spindle formation and promotes cell division. AURKA expression is increased in PCa, and its level is associated with tumor aggressiveness [11–14]. Early genetic studies of Aurora A mutants consistently revealed defects in the formation and regulation of the bipolar spindle during mitosis. A more in-depth examination of Aurora A’s expression, activation, and phosphorylation targets has further clarified its role in a series of earlier cell cycle events that set the stage for proper mitotic progression [15]. Aurora A governs several key processes, including centrosome maturation and separation, bipolar spindle assembly, mitotic entry initiation, chromosome alignment in metaphase, and cytokinesis/abscission. Additionally, the proteolytic degradation of Aurora-A is essential for cells to transition into G1 [15]. AURKA is frequently overexpressed in NEPC and CRPC, contributing to the aggressive nature of these cancers. In patients who developed treatment-induced neuroendocrine PCa, the AURKA gene is amplified in 65% of primary tumors and 86% of metastatic tumors [16, 17]. Increased AURKA not only promotes mitotic spindle formation and cell cycle progression but also interacts with other oncogenic pathways, including those involving MYCN and AR variants [16, 18]. The reciprocal regulation between AURKA and tumor suppressors like NKX3.1 further underscores its role in NEPC pathogenesis [18].
AURKA phosphorylates p53 at specific sites, leading to its destabilization and degradation. For instance, AURKA phosphorylates p53 at Ser315, which facilitates its ubiquitination by Mdm2 and subsequent proteolysis. This degradation of [19] reduces its tumor suppressor functions, including cell cycle arrest and apoptosis, thereby allowing cancer cells to proliferate despite the presence of functional p53.
Additionally, AURKA can phosphorylate p53 at Ser215, which abrogates p53’s DNA binding and transactivation activity, further impairing its ability to induce the expression of downstream target genes such as p21 and PTEN [20]. This inhibition of p53’s transcriptional activity contributes to the survival and proliferation of cancer cells under therapeutic pressure.
Moreover, AURKA disrupts the interaction between p53 and its coactivators, such as hnRNPK, by phosphorylating hnRNPK, which further diminishes p53’s transcriptional activity [21]. This multifaceted inhibition of p53 by AURKA allows cancer cells to bypass p53-mediated growth suppression and apoptosis, contributing to drug resistance.
In the context of NEPC, AURKA overexpression is often observed and is associated with the aggressive behavior of these tumors. The ability of AURKA to inactivate p53 through multiple mechanisms underscores its role in overcoming drug resistance and promoting NEPC progression [22, 23].
In a recent study, Gritsina et al. reported that C-X-C chemokine receptor type 7 (CXCR7) is upregulated in NEPC [24], following up their earlier study demonstrating a causative role of CXCR7 in driving enzalutamide-resistant PCa and thus, CRPC progression [25]. They showed that CXCR7 promotes tumor growth and proliferation by activating downstream signaling pathways, such as AURKA [24]. Specifically, CXCR7 recruits β-arrestin (ARRB2) and forms a complex that activates AURKA, a key regulator of mitosis. They confirmed in PCa patient datasets that AURKA signal transduction is positively correlated with CXCR7 expression. Further, the study tested the AURKA inhibitor alisertib and revealed that alisertib abolishes CXCR7-driven PCa growth in cell lines as well as animal models [24]. The authors thus proposed AURKA as a critical therapeutic target for CXCR7-high CRPC and NEPC patients. However, a Phase II clinical trial of alisertib in CRPC/NEPC patients failed to meet its primary endpoint due to drug toxicity and patient variability despite significant clinical benefits in a small subset of four patients with high AURKA expression [26]. Further evaluation of the drug might benefit from molecular biomarkers to preselect patients with AURKA activation and/or drug combinations to lower the dose and reduce toxicity. Alternative AURKA inhibitors are currently being investigated. Some of these include the evaluation of AL8326 in ≥2nd line small cell lung cancer, a study of Tinengotinib (TT-00420) in combination with standard treatments in people with prostate cancer, and JAB-2485 activity in adult patients with advanced solid tumors [27–29]. Notably, inhibiting AURKA is synthetically lethal with the loss of RB1 or p53 tumor suppressor genes due to AURKA’s vital role in driving the cell cycle in these tumors [30, 31]. Likewise, cells harboring a high mutational burden, as often seen in tumors with defective homologous recombination (HR), might become increasingly reliant on AURKA activity (Figure 1). In these cells, AURKA might help bypass cell cycle checkpoints, thereby enabling continued replication and tumor growth despite DNA damage [32]. As such, AURKA activity has been closely linked to tumorigenesis and the expression of genes associated with metastasis [33]. Inhibition of AURKA activity has been shown to cause delayed mitotic progression, mitotic failure, and ultimately, cell death [33]. These findings suggest that AURKA could serve as a promising therapeutic target, particularly in tumors with AURKA amplification or up-regulation, such as CRPC and NEPC. Furthermore, AURKA overexpression may contribute to resistance to therapies that target DNA repair pathways, such as PARP inhibitors like Olaparib, as well as resistance to androgen receptor pathway inhibitors (ARPi) and chemotherapy [34]. AURKA may be essential for driving cell cycle progression in PCa cells exposed to DNA-damaging therapeutics, including PARPi, ARPi, and chemotherapeutic agents [34]. To this end, it will be essential to evaluate the frequency of genomic alterations, including AURKA amplification or overexpression, RB1 loss and HR gene mutations in CRPC and NEPC tumors, and their association with therapeutic (PARPi, ARPi, and Chemotherapy) responses. If positive correlations were discovered, it would support targeting AURKA to overcome therapeutic resistance and improve treatment outcomes for these aggressive and treatment-refractory cancers.
In summary, prostate cancer progresses to CRPC through mechanisms that sustain AR signaling despite androgen deprivation. A subset of CRPC tumors further evolves into NEPC, characterized by AR independence and neuroendocrine differentiation, driven by genetic and epigenetic alterations [1, 4, 35]. In prostate cancer and CRPC with neuroendocrine traits, CXCR7 activates AURKA, promoting NEPC growth. Targeting AURKA, either directly or through its upstream regulators like CXCR7, represents a promising therapeutic strategy for managing NEPC. AURKA is overexpressed in PCa, particularly in CRPC and NEPC, where it promotes cell division, tumor aggressiveness, and metastasis. Targeting AURKA may provide a promising therapeutic strategy, especially in combination with PARPi like Olaparib, to improve outcomes in HR-deficient PCa, including cases with mutations beyond BRCA1/2. Similarly, AURKA inhibition in patients with wild-type HR genes could be utilized, as it has been shown to regulate the stability of Myc proteins, which in turn influence the expression of HR-related genes. This strategy could enhance the effectiveness of PARPi in HR-proficient PCa, particularly in tumors with high Myc expression, by inducing vulnerabilities similar to those seen in BRCA-mutant cancers. Last, AURKA inhibitors might improve outcomes of standard-of-care of metastatic PCa that uses AR pathway inhibitors, such as Enzalutamide, by intensifying AR inhibition, increasing DNA-damage-related cell death, and preventing the development of escape mechanisms, such as AR variants.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Feng Q, He B: Androgen receptor signaling in the development of castration-resistant prostate cancer. Frontiers in oncology 2019, 9:858.31552182 10.3389/fonc.2019.00858 PMC 6738163 · doi ↗ · pubmed ↗
- 2Fontana F, Limonta P: Dissecting the hormonal signaling landscape in castration-resistant prostate cancer. Cells 2021, 10(5):1133.34067217 10.3390/cells 10051133 PMC 8151003 · doi ↗ · pubmed ↗
- 3Coutinho I, Day TK, Tilley WD, Selth LA: Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence. Endocrine-related cancer 2016, 23(12): T 179–T 197.27799360 10.1530/ERC-16-0422 · doi ↗ · pubmed ↗
- 4Imamura J, Ganguly S, Muskara A, Liao RS, Nguyen JK, Weight C, Wee CE, Gupta S, Mian OY: Lineage plasticity and treatment resistance in prostate cancer: the intersection of genetics, epigenetics, and evolution. Front Endocrinol (Lausanne) 2023, 14:1191311. doi:10.3389/fendo.2023.1191311.37455903 PMC 10349394 · doi ↗ · pubmed ↗
- 5Maylin ZR, Smith C, Classen A, Asim M, Pandha H, Wang Y: Therapeutic Exploitation of Neuroendocrine Transdifferentiation Drivers in Prostate Cancer. Cells 2024, 13(23):1999.39682746 10.3390/cells 13231999 PMC 11639977 · doi ↗ · pubmed ↗
- 6Nie J, Zhang P, Liang C, Yu Y, Wang X: ASCL 1-mediated ferroptosis resistance enhances the progress of castration-resistant prostate cancer to neurosecretory prostate cancer. Free Radical Biology and Medicine 2023, 205:318–331. doi:10.1016/j.freeradbiomed.2023.06.006:37355053 · doi ↗ · pubmed ↗
- 7Rodarte KE, Nir Heyman S, Guo L, Flores L, Savage TK, Villarreal J, Deng S, Xu L, Shah RB, Oliver TG, : Neuroendocrine Differentiation in Prostate Cancer Requires ASCL 1. Cancer Res 2024, 84(21):3522–3537. doi:10.1158/0008-5472.Can-24-1388.39264686 PMC 11534540 · doi ↗ · pubmed ↗
- 8Tang F, Xu D, Wang S, Wong CK, Martinez-Fundichely A, Lee CJ, Cohen S, Park J, Hill CE, Eng K: Chromatin profiles classify castration-resistant prostate cancers suggesting therapeutic targets. Science 2022, 376(6596):eabe 1505.35617398 10.1126/science.abe 1505 PMC 9299269 · doi ↗ · pubmed ↗