Engineering Saccharomyces cerevisiae to improve heterologous abscisic acid production
Maximilian Otto, Sara Muñiz-Calvo, Michael Gossing, Florian David, Verena Siewers

TL;DR
Researchers engineered yeast to produce more abscisic acid, a plant hormone with agricultural and medical uses, by optimizing enzyme activity and using plant proteins.
Contribution
A novel strategy using plant proteins to enhance P450 monooxygenase activity in yeast for improved abscisic acid production.
Findings
Overexpression of Arabidopsis thaliana proteins AtMSBP1 and AtCOL4 increased ABA titers by over fivefold.
Knockout of PAH1 improved ABA production but caused growth defects, which were mitigated by promoter replacement.
Heterologous plant proteins outperformed complex genetic modifications in ABA titer improvement.
Abstract
Abscisic acid (ABA) is a phytohormone involved in regulating plant growth, development, and stress responses. Its various physiological activities in plants and animals make the molecule a high-value product with agricultural, medical and nutritional applications. We previously constructed an ABA cell factory by expressing the ABA metabolic pathway from Botrytis cinerea in the biotechnological workhorse Saccharomyces cerevisiae. In this study, we aimed to improve ABA production and explored various rational engineering targets mostly focusing on increasing the activity of the two cytochrome P450 monooxygenases of the ABA pathway, BcABA1 and BcABA2. We evaluated the effects of cell membrane transporters, expression of heterologous cytochrome b5, improving heme supply, altering ER homeostasis, expression of Arabidopsis thaliana proteins and improving the precursor supply. One of the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
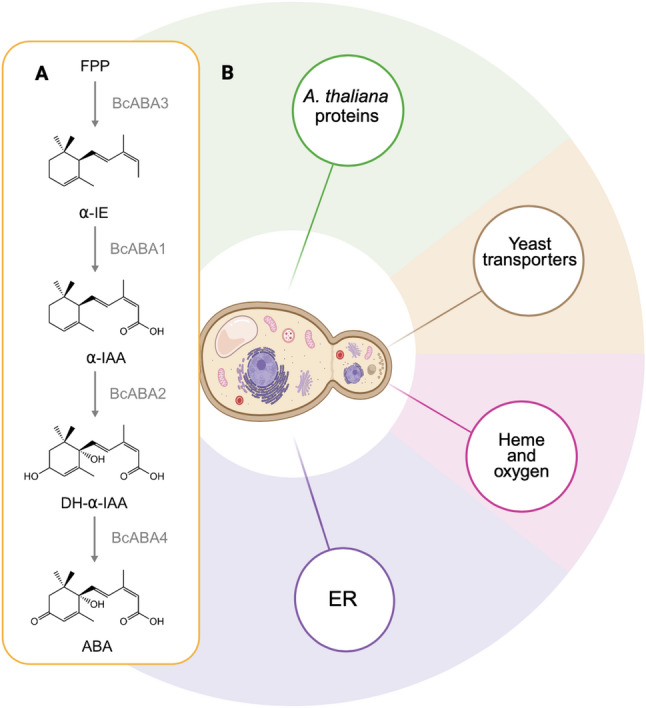 Figure 1
Figure 1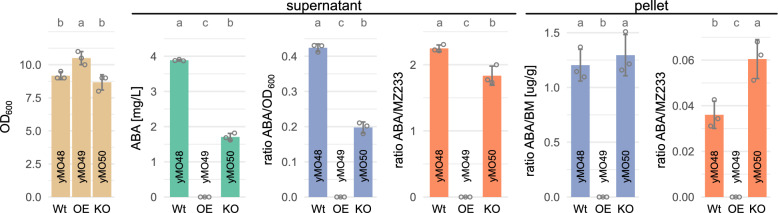 Figure 2
Figure 2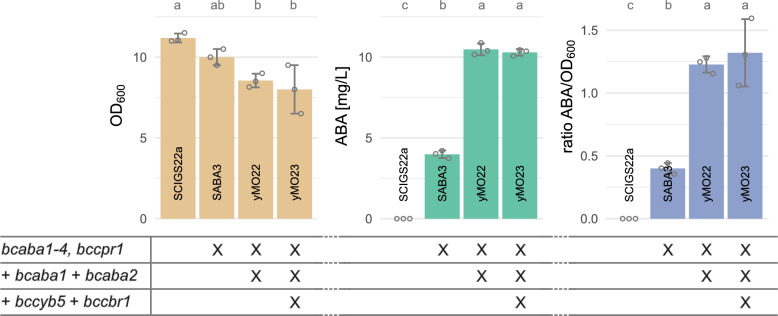 Figure 3
Figure 3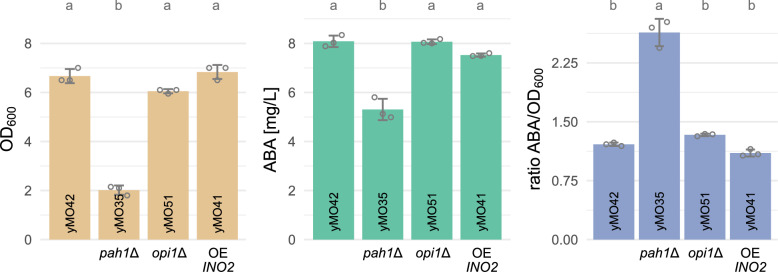 Figure 4
Figure 4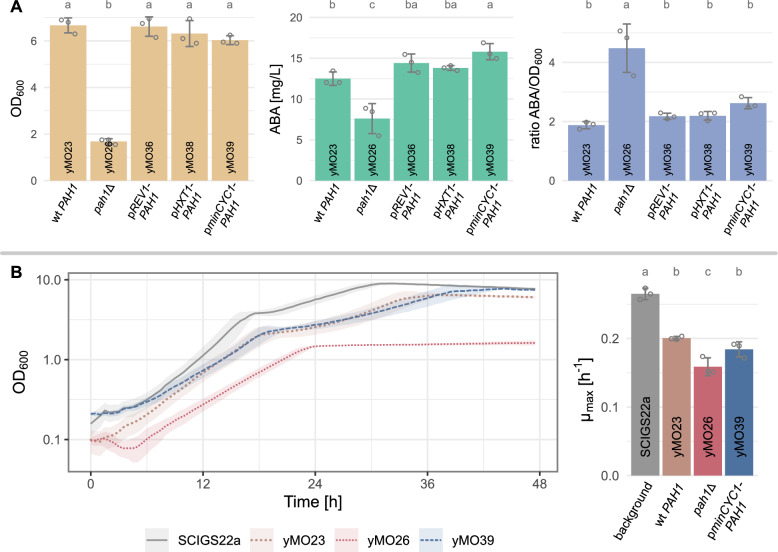 Figure 5
Figure 5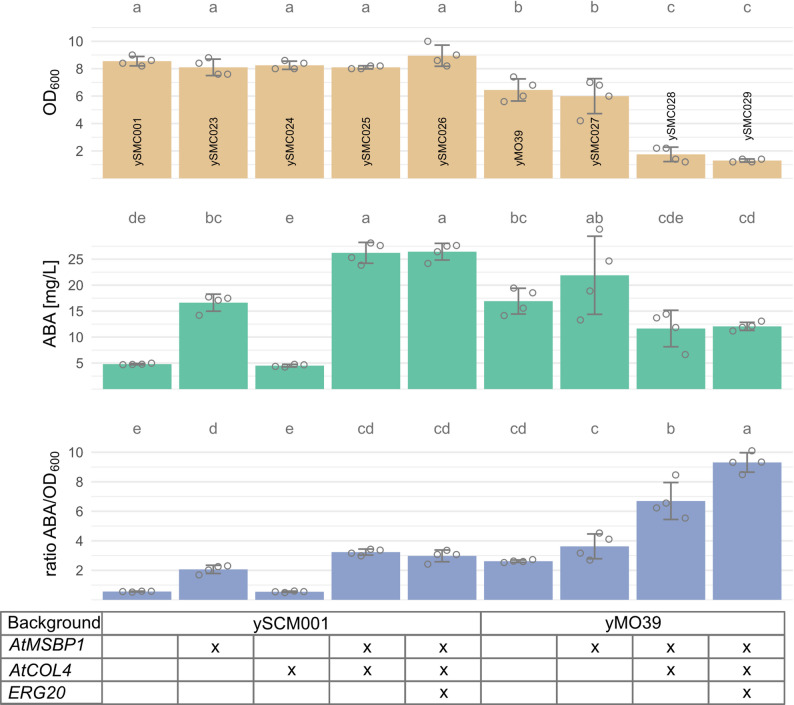 Figure 6
Figure 6- —Chalmers University of Technology
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Stress Responses and Tolerance · Plant Gene Expression Analysis · Plant Molecular Biology Research
Background
The isoprenoid abscisic acid (ABA) has become a molecule-of-interest for a multitude of applications in agriculture and medicine as well as nutrition. Its central role in plant physiology has long been known and its complex physiological interactions are still being investigated [1, 2]. Agricultural applications of ABA range from alleviating various abiotic stresses to regulating seed dormancy, germination and fruit ripening [3]. ABA can also act as a signalling molecule in organisms other than plants [4]. In recent years, ABA was shown to have various biological activities in mammals, making it a promising drug candidate. For example, pharmacological activity against various inflammatory diseases, pathogen-mediated infections, type-2-diabetes and metabolic syndrome has been reported [5–7]. ABA was furthermore identified as a bitter receptor blocker with potential applications in nutritional products [8].
A sustainable and inexpensive product source will be required to utilise ABA in these applications. The plant pathogenic fungus Botrytis cinerea is a natural producer of ABA and has been used as a biotechnological production host [9]. In B. cinerea, ABA is produced via the mevalonate (MVA) pathway from farnesyl-pyrophosphate (FPP) [10]. FPP is cyclised by the enzyme BcABA3 and subsequently oxidised at multiple positions by BcABA1, BcABA2 and BcABA4 to form ABA (Fig. 1A) [11–14]. However, the shortage of genetic tools for B. cinerea, as well as its hyphal morphology and comparatively slow growth make the fungus a challenging cell factory.Fig. 1. Overview of the engineered ABA production in S. cerevisiae. A ABA pathway in Botrytis cinerea [14]. The four pathway genes bcaba1, bcaba2,* bcaba3* and bcaba4, as well as the B. cinerea cytochrome P450 reductase encoding gene bccpr1 were expressed in yeast to enable heterologous ABA production from the native precursor FPP [15]. B Schematic representation of different strategies explored in this study to enhance ABA production. Image created with BioRender.com. FPP farnesyl pyrophosphate, α-IE α-ionylideneethane, α-IAA α-ionylideneacetic acid, DH-α-IE 1′,4ʹ-trans-dihydroxy-α-ionylideneacetic acid, ABA abscisic acid, ER endoplasmic reticulum
Saccharomyces cerevisiae has long been a workhorse for biotechnological applications. Its capabilities for high-level production of sesquiterpenes or sesquiterpenoids have been demonstrated previously, e.g. for the biofuel farnesene [16] or the anti-malarial drug precursor artemisinic acid [17]. In a previous study, we established a S. cerevisiae ABA cell factory by introducing the ABA pathway enzymes BcABA1, BcABA2, BcABA3 and BcABA4 as well as the B. cinerea cytochrome P450 reductase BcCPR1 [15]. We demonstrated that two cytochrome P450 monooxygenases (CYPs, putatively of class II), BcABA1 and BcABA2, are limiting ABA titers in the current strain [15]. This pathway bottleneck was also observed in following studies [18–20].
CYPs are a large superfamily of enzymes that catalyse oxidation reactions using molecular oxygen and contain heme as a co-factor [21]. They are ubiquitous in plant and microbial biosynthetic pathways and have become a major engineering target in biotechnology [22, 23]. Class II CYP enzymes, the most common class in eukaryotes, require cytochrome P450 reductases (CPRs) as co-enzymes for the transfer of electrons from NADPH [24]. It was also shown that co-expression of cytochrome b5 (CYB5) and the cognate reductase (CBR) can have a positive impact on CYP activities, most likely by acting as additional electron donors [17, 25, 26].
Eukaryotic CYPs, CPRs, CYB5s and CBRs are usually anchored in the ER membrane which poses additional engineering challenges. It is presumed that available membrane space and/or its lipid composition significantly affect enzyme abundance and activity in the cell [22, 27]. Therefore, modulation and expansion of the ER membrane, also referred to as ER proliferation, has become a common engineering strategy for membrane-associated enzymes like CYPs and their co-enzymes. Various native target genes, most commonly PAH1, INO2 and OPI1, have been investigated regarding ER proliferation, some of which led to multi-fold titer increases [28, 29]. PAH1 encodes phosphatidate phosphatase, a highly regulated enzyme catalysing the conversion of phosphatidates to diacylgylcerols, a key reaction for balancing levels of membrane phospholipids (PL) and triacylglyceride (TAG) storage lipids [30]. Ino2 and Opi1 are transcription factors regulating various lipid metabolism genes. The Ino2/Ino4 complex activates genes involved in PL biosynthesis, whereas Opi1 acts as a repressor when interacting with Ino2 [31]. Knockout of PAH1 or OPI1 and overexpression of INO2 result in similar phenotypes with an enlarged ER [28, 29, 32]. However, the effect of individual target genes appears to be strain- or product-specific.
Other strategies to optimize CYP activity include improving heme supply (an essential co-factor of CYPs, CPRs and CYB5s) or improving the availability of dissolved oxygen [20, 33, 34]. Overexpression of coproporphyrinogen III oxidase gene HEM13 and deletion of the heme oxygenase gene HMX1 appear to be promising targets to increase heme production [35]. Heterologous expression of the bacterial hemoglobin gene from Vitreoscilla stercoraria, vsvhb, has been investigated before with the goal of enhancing oxygen uptake and utilization in cell factories [36, 37]. Vsvhb expression furthermore benefited cell growth and protein synthesis in a variety of hosts [38].
Jiang and co-workers [39] adopted yet another approach to improve the activity of heterologous CYPs. By screening a cDNA library of Arabidopsis thaliana, they identified genes that positively affected two different CYPs, one involved in betaxanthin production, and one involved in α-santalol production. Specifically, the membrane steroid binding protein 1 (AtMSBP1), the CONSTANS-like protein 4 (AtCOL4) and the glycine-rich RNA-binding protein 7 (AtGRP7) proved to be beneficial. AtMSBP1 is localized in the ER membrane and is suggested to have a chaperone-like function [40, 41]. AtMSBP1 also physically interacts with three monolignol biosynthetic CYPs, forming a complex on the ER membrane also known as a metabolon, which regulates the lignin biosynthetic process [40]. This organization presumably enhances enzyme stability and activity, for example through substrate channelling. AtCOL4 encodes a nuclear-localized protein that acts as a transcriptional repressor of flowering genes [42]. AtGRP7 encodes a glycine-rich RNA-binding protein that can facilitate alternative splicing for various mRNAs. It is involved in physiological processes such as promoting flowering, enhancing the innate immune system and stress response [43]. How AtCOL4 and AtGRP7 influence CYP activity is so far not elucidated.
Another engineering approach that is not directly related to CYPs but that was proven worthwhile in other cell factories that produce acidic compounds is modulating transporter expression. When produced in yeast, ABA is predominantly found in the cell culture supernatant [15]. As a weak organic acid with a pKa of 4.75 [44], ABA is mostly present as the conjugate base in the cytosol of plants, with the membrane permeability of the ion being much lower than the protonated form [45, 46]. In yeast, ABA is presumably exported by an unspecific transporter. Complex regulatory networks mediate resistance to weak organic acids in S. cerevisiae with a multitude of transporters being involved in this process [47]. Upregulation of native transporters has been investigated for other cell factories with the goal of mediating product-related cell stress, such as increased turgor pressure or oxidative stress [47]. Nonetheless, downregulation of transporters could also be a valid engineering target by preventing the export of pathway intermediates, thereby increasing their local concentration in the cytosol. Identifying transporters involved in the import or export of a given heterologous compound is challenging. An alternative approach is to overexpress or knock-out global regulators involved in transporter expression. The transcription factors Pleiotropic Drug Resistance 1 (Pdr1) and Yeast Reveromycin-A Resistant 1 (Yrr1) are two such global regulators, involved in the pleiotropic drug resistance signalling network [48, 49]. Modulation of the PDR regulatory network could provide valuable insight for ABA-producing yeast strains and other S. cerevisiae cell factories producing weak organic acids.
Our previous proof-of-concept study demonstrated the feasibility of an S. cerevisiae cell factory, but ABA titres remained low. In this study, we aimed to overcome this limitation through rational engineering. We explore a variety of approaches including the modification of native gene targets and expression of heterologous proteins (Fig. 1B).
Methods
Plasmid construction
For PCR reactions, PrimeSTAR HS DNA Polymerase (Clontech), Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific) or SapphireAmp (Takara Bio) were used, and the manufacturer’s instructions were followed. Primer sequences can be found in Supplementary Table S1 (Additional file 1) and specific conditions of standard PCR reactions are listed in Supplementary Table S2 (Additional file 1). For plasmid and PCR product purification, GeneJet Purification Kits (Thermo Fisher Scientific) were used. Sanger DNA sequencing was performed by Eurofins Genomics. Primers and other oligonucleotides were ordered from Eurofins Genomics or Integrated DNA Technologies.
Plasmids used in this study are listed in Table 1. For construction of pMG138 the backbone of pCfB2312 was amplified by PCR using primers F-HR fwd and R-HR rev. The gRNA expression cassettes targeting YRR1 and PDR1 were ordered as gene fragments. Subsequently, pMG138 was constructed by Gibson assembly using the PCR product and the gene fragment.Table 1. Plasmids used in this studyPlasmid nameUseBackboneExpression cassetteReferencep416TEFEpisomal expression–Empty[50]p413TEFEpisomal expression–Empty[50]pRS316 + prTEF1 + HEM13 + terADH1Episomal expressionpRS316pTEF1-HEM13-tADH1[35]pPM28Episomal expressionpRS316pTDH3-eroGFP*-tCYC1*[51]pSPGM-INO2Episomal expressionpSP-GM1pTEF1-INO2-tADH1[52]p423-INO2(L119A)Episomal expressionp423 + TDH3pTDH3-INO2(L119A)-tCYC1This studyp426-ICE2Episomal expressionp426 + TDH3pTDH3-ICE2*-tCYC1This studypCfB2904-ABA1-CPRGenomic integration in XI-3pCfB2904pPGK1*-bcaba1-tADH1 pTEF1-bccpr1-tCYC1[15]pCfB2909-ABA2-ABA4Genomic integration in XII-5pCfB2909pPGK1-bcaba2-tADH1 pTEF1-bcaba4-tCYC1[15]pCfB3035-ABA3Genomic integration in X-4pCfB3035pTEF1-bcaba3-tCYC1[15]pX3-bcaba1 + 2Genomic integration in X-3pMC-X3pTDH3-bcaba1-tTDH1 pCCW12-bcaba2-tENO2This studypXII2-bccyb5 + cbr1Genomic integration in XII-2pMC-XII2pHHF2-bccyb5-tPGK1 pTEF2-bccbr1-tSSA1This studypX2-atcol4Genomic integration in X-2pMC-X2pHHF2-AtCOL4-tPGK1This studypX2-msbp1Genomic integration in X-2pMC-X2pTEF2-AtMSBP1-tENO2This studypX2-atcol4/atmsbp1Genomic integration in X-2pMC-X2pHHF2-AtCOL4-tPGK1 pTEF2-AtMSBP1-tENO2This studypXI2-vsvhbGenomic integration in XI-2pMC-XI2pCCW12-vsvhb-tTDH1This studypXI5-ERG20Genomic integration in XI-5pMC-XI5pTEF2-ERG20-tTDH1This studypCfB2312Cas9 expressionpTEF1-Cas9-tCYC[53]pCfB3020gRNA expression for X-2 integration[53]pCfB3041gRNA expression for X-3 integration[53]pCfB3044gRNA expression for XI-2 integration[53]pCfB3046gRNA expression for X-5 integration[53]pCfB3051gRNA expression for X-3, XI-2, XII-2 integration[53]pMG138gRNAs expression targeting PDR1 and YRR1pCfB2312pTEF1-Cas9-tCYC1pSNR52-gRNA-tSUP4This studypWS172/pWS158Cas9 expression and gRNA expression targeting HIS3, IRE1, INO2, HMX1, HAC1pWS158 (Addgene ID: 90517)pWS172 (Addgene ID: 90519)pPGK1-Cas9-tPGK1**ptRNAPhe-gRNA-tSNR52This studypMEL10gRNA expression backbone used for knockout of PAH1 and OPI1pSNR52-gRNA-tCYC1[54]
The Golden Gate-based MoClo workflow [55–57] was followed to construct pX3-bcaba1 + 2, pXII2-bccyb5 + cbr1, pX2-atcol4, pX2-atmsbp1, pX2-atcol4 + atmsbp1, pXI2-vsvhb and pXI5-ERG20. All MoClo assemblies are listed in Supplementary Table S3 (Additional file 1). First, primers pairs 306/357 and 307/310 were used to remove a BsaI site from bcaba1 and to attach MoClo type-3 compatible overhangs via PCR. The resulting PCR products were fused together via a 2-step PCR reaction. In the first reaction, the PCR products were mixed in a 1:1 ratio (≈100 ng) with PrimeStar PCR master mix but without primers (55 °C T_ann_, 1:15 min t_elo_, 15 cycles). In the second reaction, 2 µL of the first reaction was used as template with the primer pair 306/307 (55 °C T_ann_, 1:40 min t_elo_, 35 cycles). The PCR product of the second reaction was purified and sequence verified. Primer pairs 308/309 were used to attach type-3 MoClo overhangs to bcaba2. DNA sequences lacking BsaI, BsmbI and NotI sites for bccyb5 (UniProt identifier A0A384K1M2), bccbr1 (UniProt identifier A0A384JNH0), AtCOL4 (Uniprot identifier Q940T9), AtMSBP1 (UniProt identifier Q9XFM6) and vsvhb (UniProt identifier P04252) were codon-optimised for yeast and provided by Genscript Biotech Corp. Primer pairs 292/293 and 294/295 were used to attach MoClo type-3 overhangs to bccyb5 and bccbr1, respectively.
The MoClo compatible bcaba1, bcaba2, bccyb5 and bccbr1 fragments were inserted into the MoClo entry vector pYTK001 [56] (Supp. Table S3; Additional file 1). For AtCOL4, AtMSBP1 and vsvhb, the strong Kozak sequence (TATACA) and type-3b overhangs were already included in the respectively cloning vector (pUC57-BsaI and BsmBI-free) containing each gene. Primer pairs oSMC 35/36, 37/38 and 39/40 were used to attach MoClo type-3a overhangs to the HHF2, TEF2 and CCW12 promoters, respectively.
Subsequently, level-1 MoClo plasmids were assembled for each gene. The level-1 plasmids were combined with the backbones pMC-X3, pMC-XII2. pMC-X2 and pMC-XI2 [57] to form pX3-bcaba1 + 2, pXII2-bccyb5 + cbr1, pX2-atcol4, pX2-atmsbp1, pX2-atcol4 + atmsbp1 and pXI2-vsvhb respectively.
For the construction of pXI5-ERG20, genomic DNA from CEN.PK113-5D was used as a template. Primer pairs oSMC123/124 were employed to attach type-3 MoClo overhangs to ERG20. The purified DNA fragment was then combined with pYTK014, pYTK056, and the preassembled level-2 MoClo backbone, pMC-XI5 [57], to create pXI5-ERG20 (Supp. Table S3; Additional file 1). To generate p426GPD-INO2 (L119A) and p426-ICE2, *INO2 *(L119A) was amplified from genomic DNA using the primer pair oSMC 25/26 with the template being the PCR product obtained from the colony that was successfully mutated and prior to the plasmid curation process, as is explained in the results. The ICE2 coding sequence was amplified from genomic DNA of CEN.PK113-11C using the primer pair oSMC 27/28. Both obtained PCR products were digested with the restriction enzymes XmaI and XhoI before being ligated into the corresponding restriction sites in p423GPD and p426GPD plasmids [50]. Primer pair oSMC 78/84 was used for sequencing and validation of the cloning of p423-INO2 (L119A) and p426-ICE2.
Microorganisms and media
Plasmids were used to transform NEB® 5-alpha competent E. coli cells (New England Biolabs). E. coli was cultivated at 37 °C in liquid lysogeny broth (LB) medium or on LB agar plates with appropriate antibiotics. S. cerevisiae strains are listed in Table 2 and a pedigree chart is shown in Supplementary Figure S1 (Additional file 1). E. coli was cultivated at 37 °C in liquid lysogeny broth (LB) medium or on LB agar plates with appropriate antibiotics. Yeast was cultivated at 30 °C in liquid yeast extract peptone dextrose (YPD) medium or mineral medium (adapted from [58]) while shaking at 220 rpm. For cultivation on agar plates YPD medium or synthetic defined (SD) medium was used. For all yeast media, 20 g/L glucose was used. Unless stated otherwise, SD medium was supplemented with 100 mg/L uracil or histidine for strains with uracil or histidine auxotrophy. Detailed media compositions can be found in Supplementary Table S4 (Additional file 1).Table 2. Strains used and constructed in this studyStrain nameParent strainGenomic modificationsPlasmidReferenceCEN.PK113-5D–MATa MAL2-8 ^c^ SUC2 ura3-52–[59], provided by P. Kötter, University of Frankfurt, GermanyCEN.PK113-11C–MATa MAL2-8 ^c^ SUC2 ura3-52 his3Δ–provided by P. Kötter, University of Frankfurt, GermanyyMG01CEN.PK113-11CpTEF1-YRR1 pTDH3-PDR1–This studyyMG02CEN.PK113-11Cyrr1Δ pdr1Δ–This studySCIGS22aCEN.PK113-5Dlpp1Δ::loxP dpp1Δ::loxPpERG9 Δ::loxP pHXT1**gdh1Δ::loxP pTEF1-ERG20pPGK1-GDH2 pTEF1-tHMG1–[60]SABA3SCIGS22apPGK1-bcaba1 pPGK1-bcaba2pTEF1-bcaba3 pTEF1-bcaba4pTEF1-bccpr1–[15]yMO22SABA3pTDH3-bcaba1 pCCW12-bcaba2–This studyyMO23yMO22pHHF2-bccyb5 pTEF2-bccbr1–This studyyMO26yMO23pah1Δ–This studyyMO35yMO26–p416TEFThis studyyMO36yMO23pPAH1Δ::pREV1–This studyyMO38yMO23pPAH1Δ::pHXT1–This studyyMO39yMO23pPAH1Δ::pminCYC1–This studyyMO40yMO23–pRS316 + prTEF1 + HEM13 + terADH1This studyyMO41yMO23–pSPGM-INO2This studyyMO42yMO23–p416TEFThis studyyMO48SABA3–p416TEFThis studyyMO49yMG01pPGK1-bcaba1 pPGK1-bcaba2pTEF1-bcaba3 pTEF1-bcaba4pTEF1-bccpr1p416TEF, p413TEFThis studyyMO50yMG02pPGK1-bcaba1 pPGK1-bcaba2pTEF1-bcaba3 pTEF1-bcaba4pTEF1-bccpr1p416TEF, p413TEFThis studyyMO51yMO23opi1Δp416TEFThis studyySMC001SABA3his3Δ–This studyySMC003CEN.PK113-11CINO2 (L119A)–This studyySMC017ySMC001ino2Δp423TDH3-INO2 (L119A) p426TDH3-ICE2This studyySMC019ySMC001ire1Δ HAC1ipPM28This study ySMC020ySMC001hmx1ΔpRS316 + prTEF1 + HEM13 + terADH1This studyySMC023ySMC001pTEF2-AtMSBP1–This studyySMC024ySMC001pHHF2-AtCOL4–This studyySMC025ySMC001pHHF2-AtCOL4 pTEF2-* AtMSBP1*–This studyySMC026ySMC025pTEF2-ERG20–This studyySMC027yMO39pTEF2-AtMSBP1–This studyySMC028yMO39pHHF2-AtCOL4 pTEF2-* AtMSBP1*–This studyySMC029ySMC028pTEF2-ERG20–This studyySMC030ySMC001pCCW12-vsvhb–This study
Strain construction
Yeast strains were transformed according to the protocol by Gietz and Woods [61]. The plasmids pX3-bcaba1 + 2, pXII2-bccyb5 + cbr1, pX2-atcol4, pX2-atmsbp1, pX2-atcol4 + atmsbp1, pXI2-vsvhb and pXI5-ERG20 were used for genomic integrations. The procedure described in Jessop‐Fabre et al. [53] was followed, using the Cas9-encoding plasmid pCfB2312 and the gRNA-helper plasmids pCfB3020, pCfB3041, pCfB3044, pCfB3046 and pCfB3051.
For the PDR1 and YRR1 deletion or overexpression, gRNAs were designed using Benchling. These gRNAs targeted sites upstream of the start ATG of the respective ORF, so that the same gRNA could serve for both deletion and overexpression purposes. The repair fragments for deletion of YRR1 and PDR1 were ordered as 120-bp oligonucleotides (Supp. Table S5, Additional File 1). Hybridized fragments were used along with pMG138 to transform CEN.PK113-11C. For promoter replacement, the promoter sequences pTDH3 and pTEF1 were amplified from genomic DNA of CEN.PK113-11C using primers pTDH3-PDR1 fwd + rev and pTEF1-YRR1 fwd + rev, respectively. Homology to PDR1 and YRR1 was introduced by reamplifying pTDH3 and pTEF1 with primers pTDH3-PDR1 ampl OL fwd + rev and pTEF1-YRR1 ampl OL fwd + rev, respectively. Purified fragments were used along with pMG138 to transform CEN.PK113-11C. Obtained clones were verified by colony PCR using primers PDR1 fwd + rev and YRR1 fwd + rev, respectively.
PAH1 and OPI1 were deleted following the protocol by Mans et al. [54], using the plasmid pMEL10 for gRNA expression and the Yeastriction tool (http://yeastriction.tnw.tudelft.nl/). The sequences of the gRNA fragments and repair oligos can be found in Supplementary Table S5 (Additional file 1). Primers pairs 366/367 and 1260/1261 (Supp. Table S1 and Supp. Table S2; Additional file 1) were used to validate the pah1Δ and opi1Δ genotype, respectively.
For the PAH1 promoter replacement, the gRNA was designed using the Benchling CRISPR Guide tool (Supp. Table S5) and its coding sequence inserted into pMEL10. For construction of the linear promoter replacement cassettes, primer pairs 417/418, 422/423, 424/425 were used. The primers were designed to contain homologous regions for replacing 771 base pairs upstream of the PAH1 start codon. The resulting PCR products were used as repair fragments in a transformation according to Mans et al. [54]. Primers 426/427 were used to validate the pPAH1 replacement. The colony PCR product of strain yMO36 was sequenced for verification.
To delete HIS3, IRE1, INO2 and HMX1, the protocol described by Shaw et al. [62] was used. Briefly, two oligonucleotides encoding the gRNA were annealed in vitro and integrated into the CRISPR/Cas9 expression plasmid pWS158 or pWS172 using a BsmBI Golden Gate assembly. Primer pairs oSMC: 57/58, 29/30, 1/2 and 48/49, were used to generate sgRNA for HIS3, IRE1, INO2 and HMX1, respectively.
The constructed plasmids pWS158 or pWS172were used to transform the respective yeast strain together with the donor DNA to facilitate homology-directed repair at the double-strand break. Donor DNA was created by no-template PCR of partially overlapping primers, consisting of the 60 nucleotides upstream of the start codon, followed by the 60 nucleotides downstream of the stop codon, as previously described [63]. The DNA sequences can be found in Supplementary Table S5 (Additional file 1). Primer pairs oSMC: 89/90, 11/14, 5/6 and 59/60 were used were used to validate the his3Δ, ire1Δ, ino2Δ and hmx1Δ genotypes, respectively.
To introduce the point mutation L119A in INO2, CEN.PK113-11C was transformed with pWS158 expressing a gRNA targeting the locus and the donor DNA containing the desired mutation (together with a synonymous mutation in the target sequence to prevent Cas9 cleavage after repair) [63]. Primer pairs oSMC 1/2 and oSMC 5/6 were used to generate gRNA and for sequence validation, respectively.
To obtain the HAC1i genotype, the gRNA-Cas9 expression plasmid pWS172 was cotransformed with the donor DNA. The gRNA targeting the HAC1 intron was generated by annealing the oligos oSMC33 and oSMC34. The donor DNA for repair consisted of the spliced HAC1 version (HAC1i), obtained by fusing two DNA fragments of HAC1 using primer pairs oSMC 15/16 and oSMC 17/18 through overlap extension PCR. Primers oSM31 and oSM32 were used to validate the intron deletion by colony PCR and sequencing.
Codon-optimized sequences of heterologous genes used in this study can be found in Supplementary Table S6 (Additional file 1).
Cultivation for ABA analysis
Single colonies were picked from agar plates for precultures (1.5 mL mineral media) in 14 mL round-bottom cultivation tube (Greiner Bio-One). Precultures were grown for 48 or 72 h (for slow growing pah1Δ strains) and main cultures (2.5 mL mineral media) were inoculated at OD_600_ 0.1 in 24-deepwell microplates (square wells, pyramid-bottom, EnzyScreen). Cultures were grown for 48 h (Figs. 3, 4) or 60 h (Fig. 5), OD_600_ was measured, cultures were centrifuged (5 min, 1500×g) and 1 mL supernatant was transferred to 2-mL Eppendorf tubes.
ABA extraction and quantification
For Figs. 2, 3, 4 and 5, ABA was extracted similarly to the procedure described in [15]. One mL of ethyl acetate (>99.9% Sigma-Aldrich) containing 0.5% (v/v) formic acid (>98%, Sigma-Aldrich) was added to the 2-mL Eppendorf tubes containing 1 mL culture supernatant. The tubes were vortexed for 10 s, then centrifuged (10 min, 13,000×g, 4 °C), and 0.8 mL of the supernatant was transferred to a new Eppendorf tube. The solvent was evaporated using a Genevac miVac (45 min, 20 mBar, 45 °C). The pellet was reconstituted in 0.8 mL methanol (>99.9%, Sigma-Aldrich), centrifuged (10 min, 13,000×g, 4 °C) and ≈0.5 mL of the supernatant was transferred to an HPLC (high-performance liquid chromatography) vial.
A simpler sample preparation procedure was used for the experiments displayed in Fig. 6 and Supplementary Figures S4 (B, C) and S5 (Additional file 1). Cell cultures were centrifuged (5 min, 2000×g), then 1 mL of supernatant was filtered using a 0.22‐µm nylon filter and transferred to an HPLC vial.
The samples were analysed in an Agilent 6120 Single Quadrupole mass spectrometer (MS) with an Agilent Infinity 1260 HPLC system consisting of a binary pump, autosampler and thermostat column compartment. Ionization was performed using an atmospheric pressure electrospray ionization (API-ES) source (positive mode). Compounds were separated on an Agilent Poroshell 120 EC-C18 (2.7 μm, 3.0 × 50 mm) column (maintained at 40 °C) using a water-acetonitrile gradient with 0.04% formic acid in both solvents. The gradient started with 95% water and, over 5 min, gradually changed to 95% acetonitrile (> 99.5%, Sigma-Aldrich). After a 2-min hold, the gradient was ramped back to 95% water over 3 min. The sample injection volume was set to 10 µL. ABA was quantified by selected ion monitoring (m/z = 265) and (S)-(+)-ABA standard (>98%, Cayman Chemicals) was used to fit a calibration curve (2nd polynomial).
Growth Profiler analysis
Growth (Fig. 5B) was analysed using a Growth Profiler 960 (Enzyscreen). Precultures were prepared as described above and cultures were subsequently grown in transparent bottom 96-well plate (Enzyscreen) with 250 µL mineral media per well. Pictures were taken every 30 min.
Software and statistical analysis
Besides the manufacturer’s software for the Growth Profiler (Enzyscreen) and the HPLC–MS (Agilent), R Studio [64] was used to analyse the data. Relevant R packages include tidyverse [65], emmeans [66], multcomp [67] and growth rates [68]. Benchling (www.benchling.com) was used for planning and analysing DNA constructs. For supplementary figures S4 and S5 (Additional file 1) data were plotted using GraphPad Prism 9.5.1 (GraphPad Software).
One-way ANOVA followed by Tukey's honest significance test (⍺ = 0.05) was used to determine significant differences between the strains. Results are displayed as compact letter display above the bar charts. For two groups’ comparison of data, an unpaired student’s t-test was used.
Results and discussion
Effects of transporter expression modulation on ABA production
Overexpression or knock-down of native transporters could be beneficial for ABA production, either by alleviating product-related cell stress, as was shown for the anti-malaria drug precursor artemisinic acid [69], or by preventing the export of pathway intermediates.
To test the two hypotheses, the endogenous genes encoding two global transcriptional regulators involved in transporter expression, Yrr1 and Pdr1, were either overexpressed using strong, constitutive promoters, or deleted. Altered transporter activity in the resulting strains was confirmed by evaluating the response to the antifungal fluconazole. As expected, while overexpression of PDR1 and YRR1 resulted in increased fluconazole resistance, their deletion resulted in increased fluconazole sensitivity (Supp. Fig. S2, Additional file 1).
Next, the B. cinerea genes bcaba1, bcaba2, bcaba3, bcaba4 and bccpr1 were integrated in the strains’ genomes to establish ABA production. Figure 2 shows the measured OD_600_, ABA titer, ratio of ABA to OD_600_, and ratio of ABA to an unknown compound with the mass-to-charge (m/z) value 233.15, referred to as MZ233. MZ233 was earlier detected in the supernatant of ABA-producing strains and is assumed to be an intermediate or side-product in the ABA pathway [15]. ABA and MZ233 were quantified separately in the supernatant and cell pellet.Fig. 2. Effects of native transporter expression modulation in ABA-producing strains. Genes encoding the transcription factors Pdr1 and Yrr1 were either overexpressed (yMO49) or knocked out (yMO50) and compared to a control strain with wild-type PDR1 and YRR1 expression (yMO48). OD_600_ (beige), ABA titer (green), ABA titer normalized to OD_600_ or biomass (blue), and ion count ratio of ABA to unknown metabolite MZ233 (orange) are shown after 48 h of cultivation in mineral medium (24-deepwell microplates). Supernatant and cell pellet were analysed separately. Grey circles show the individual data points used to calculate the mean and standard deviation. Only traces of ABA (<0.05 mg/L) and MZ233 were detected for yMO49. Significance is shown as compact letter display above the bars. Shared letters indicate no significant difference. Wt wild-type, OE overexpression, KO knockout
Small, but significant differences were detected in the OD_600_ at 48 h, with yMO49, overexpressing PDR1 and YRR1, exhibiting a higher OD_600_ compared to the control, yMO48, and the double-knock-out strain, yMO50 (Fig. 2). Surprisingly, only traces of ABA and MZ233 were detected in the supernatant and pellet of the overexpression strain yMO49. About 50% less ABA was detected in the supernatant of the knock-out strain yMO50 compared to the control; however, the amount of ABA in the cell pellet remained the same. In the supernatant, the control strain showed a significantly higher ratio of ABA/MZ233 compared to yMO50, though the difference was only 20%. In contrast, the ABA/MZ233 ratio was 70% higher for yMO50 in the cell pellet. This shows that, when normalized to ABA, the double-knock-out strain contains more MZ233 in the supernatant and less MZ233 in the cell pellet.
Neither overexpression of PDR1 and YRR1, nor their deletion improved ABA production. In the overexpression strain yMO49, ABA intermediates, likely upstream of MZ233, might be exported rapidly, thereby removing substrates from the product pathway and resulting in only traces being detectable. However, no additional peaks of postulated intermediates were identified in the HPLC–MS chromatogram of the supernatant when comparing yMO49 to yMO48 (Supp. Fig. S3, Additional file 1) and high-resolution MS analysis would be necessary to confirm this hypothesis. The increase in OD_600_ for yMO49 indicates that the strain experiences less weak-acid-related stress [47, 69]. The reasons for the reduced ABA titers in yMO50 compared to the wild-type control are unclear. Nonetheless, the higher intracellular ABA/MZ233 ratio for yMO50 (Fig. 2) suggests that, ratio wise, more MZ233 is converted to ABA in yMO50 (assuming that MZ233 is a pathway intermediate). The results indicate that one or multiple Pdr1/Yrr1-regulated transporters facilitate the transport of ABA pathway intermediates—if not of the product itself. Pdr1 and Yrr1 control the expression of several genes encoding ATP-binding cassette (ABC) transporters involved in the multidrug response in yeast, such as YOR1, SNQ2, PDR5, PDR10 and PDR15 (reviewed in [70]). Modulating individual transporters could still be worth investigating in future studies. This is exemplified by a study investigated the effect of expressing A. thaliana ABA transporters in an ABA-producing Y. lipolytica strain, with the aim of reducing cellular stress; however, ABA titers remained unchanged [18]. In another recent study native ABC transporters were overexpressed or deleted in an ABA-producing Y. lipolytica strain [71]. Specifically, overexpression of the gene encoding the transporter YlGcn20 (named due to its similarities to the S. cerevisiae protein Gcn20) improved ABA production by about 10%, while the overexpression of other transporters notably decreased ABA levels and/or biomass accumulation [71]. Wang et al. [72] demonstrated how disruption of the transportome in S. cerevisiae can be used to identify native genes involved in the transport of heterologous products. The same approach could be followed for an ABA-producing strain.
Genomic integration of additional pathway gene copies and improving co-enzyme and co-factor availability
The previously engineered S. cerevisiae strain SABA3 was used as the starting strain in this study [15]. This strain is derived from the terpenoid platform strain SCIGS22a with increased flux through the MVA pathway and improved NADPH supply [60, 73], and contains single copies of the B. cinerea genes bcaba1, bcaba2, bcaba3, bcaba4, and bccpr1 (Fig. 1A; Table 2). In our previous study, we showed that plasmid-based expression of additional gene copies increased the ABA titer more than fourfold [15]. Genomic integration of expression cassettes is mostly preferred to episomal expression for biotechnological applications, since it results in less cell-to-cell variability and higher genetic stability [56, 74]. Therefore, additional copies of bcaba1 and bcaba2 were integrated into the genome of SABA3 resulting in the strain yMO22. Bcaba1 and bcaba2 were expressed using the strong promoters pTDH3 and pCCW12, respectively. Furthermore, we investigated the effect of expressing the B. cinerea cytochrome b5, encoded by bccyb5, and cognate cytochrome b5 reductase, encoded by bccbr1. The strain yMO23 (originating from yMO22) contains an expression cassette with pHHF2-bccyb5 and pTEF2-bccbr1 integrated in the genome.
As expected, the additional copies of bcaba1 and bcaba2 resulted in an increase in ABA production, both in the absolute titer and titer normalised to OD_600_ (Fig. 3). The 2.6-fold increase when comparing SABA3 and yMO22 was lower than the 4.1-fold increase that was previously observed in the plasmid-carrying strain [15]. This was anticipated since plasmid-based gene expression levels are often higher when compared to genomic integrations [56, 75], likely due to cells carrying on average more than one copy of centromeric plasmids [76]. These results indicate that CYP activity still limits ABA titers.Fig. 3. Effects of expressing additional copies of bcaba1 and bcaba2, as well as expressing the B. cinerea CYB5 (bccyb5) and its cognate CBR (bccbr1). “X” indicates presence of the genetic modification in the strain. OD_600_ (beige), ABA titer in the supernatant (green) and ABA titer normalized to OD_600_ (blue) are shown after 48 h of cultivation in mineral medium (24-deepwell microplates). Grey circles show the individual data points used to calculate the mean and standard deviation. Strain SCIGS22a not containing any ABA pathway genes was used as control. Significance is shown as compact letter display above the bars. Shared letters indicate no significant difference
Conversely, expression of bccyb5 and bccbr1 in yMO23 did not result in an increase of ABA titers (Fig. 3) which highlights the complex roles of CYB5s. For some CYPs, the co-enzyme is required for the reaction to be catalysed while in other cases CYB5s have a modulating effect (stimulatory or inhibitory) [77].
We also tested if the supply of heme is limiting productivity; however overexpression of the coproporphyrinogen III oxidase gene HEM13 did not improve ABA titers (Supp. Fig. S4A; Additional file 1) and only a slight improvement of 20% was observed when combined with the deletion of the heme oxygenase gene HMX1 (Supp. Fig. S4B; Additional file 1). We also expressed vsvhb with the goal of improving oxygen availability. However, this did not benefit ABA titer nor cell growth (Supp. Figure S4 C; Additional file 1). These data indicate that oxygen availability and heme supply is sufficient in the current strain.
Effects of ER proliferation on ABA production
ER proliferation is a promising engineering strategy for ABA cell factories since it was successfully applied for other isoprenoid-producing strains and does not require engineering of the heterologous CYP itself [28, 29]. Three of the most commonly targeted native genes for ER proliferation are PAH1, INO2 and OPI1. We investigated the effects of a PAH1 or OPI1 knockout (strains yMO35 and yMO51 respectively) and INO2 overexpression (strain yMO41). Even though the addition of bccyb5 and bccbr1 in yMO23 did not result in higher ABA titers (Fig. 3), we decided to use the strain for further experiments, since BcCYB5 activity could also be affected by ER expansion. INO2 was overexpressed using a centromeric plasmid while the other strains analysed in this experiment were transformed with an empty plasmid to render them prototrophic.
Figure 4 shows the OD_600_, ABA titer and ABA titer normalized to OD_600_ for strains with engineered PL metabolism. OD_600_ and ABA titers were comparable for all strains, with exception of the pah1Δ strain yMO35. yMO35 exhibited a severe growth defect with ≈70% lower OD_600_ after 60 h compared to the control strain yMO42. Absolute ABA titers for yMO35 were decreased by ≈35%; however, the ABA titer relative to the OD_600_ was increased 2.2-fold. The results suggest that pah1Δ-mediated ER expansion was beneficial for ABA production. However, the positive effects of the PAH1 knockout came with impaired growth, a phenotype that has been observed before [78–80].Fig. 4. Effects of pah1Δ, opi1Δ or INO2 overexpression on ABA production. Genetic modifications additional to yMO42 (yMO23 carrying an empty plasmid) are displayed below the bars and include knockout of PAH1 (yMO35) or OPI1 (yMO51) and episomal overexpression of INO2 (yMO41). OD_600_ (beige), ABA titer in the supernatant (green) and ABA titer normalized to OD_600_ (blue) are shown after 60 h of cultivation in mineral medium (24-deepwell microplates). Grey circles show the individual data points used to calculate the mean and standard deviation. Significance is shown as compact letter display above the bars. Shared letters indicate no significant difference. OE overexpression
We decided to also investigated other strategies to modulate the ER microenvironment including overexpression of ICE2 (encoding a protein indirectly involved in Pah1 regulation), de-regulation of Ino2 via an L119A point mutation and provoking the ER-based unfolded protein response (UPR) (via simultaneous knockout of IRE1, HAC1i expression and eroGFP expression) in strain ySMC019 [81–84]. Interestingly, integrating the L119A mutation in INO2 in the genome was highly unstable and reverted within 72 h (data not shown). Instead, we explored episomal expression of the L119A mutant. However, none of these modifications resulted in higher ABA titer (Supp. Fig. S5; Additional file 1). A higher ABA yield was observed for the strain with upregulated UPR, ySMC019, but the strain also exhibited a severe growth defect (Supp. Fig. S5; Additional file 1).
Even though modification of the target genes PAH1, OPI1, INO2 and ICE2 all resulted in ER expansion in other reports, they did not all affect CYP activity in the ABA-producing strains. A previous study compared the effects of pah1Δ, opi1Δ and overexpression of INO2 for the production of isoflavonoids [85]. In that study, flavonoid titers were improved by either knocking out OPI1 or by overexpressing INO2. No beneficial effects were observed for pah1Δ, and severe growth defects were only seen for a pah1/opi1 double-knock-out strain and a strain with opi1Δ and simultaneous INO2 overexpression [85]. Interestingly, another report showed that deletion of PAH1 in Y. lipolytica resulted in an improvement in ABA production, whereas INO2 overexpression or OPI1 deletion had no effect [20]. This is in accordance with our observations in S. cerevisiae (Fig. 4).
As expected from previous reports, the beneficial effects of ER proliferation are strain- and/or product-specific, potentially depending on other genetic modifications or being specific to the heterologous CYP. In this context it is noteworthy that the ABA-producing strains of this study are based on the sesquiterpenoid platform strain SCIGS22a, in which the genes DPP1 and LPP1 were deleted to prevent the dephosphorylation of farnesyl-pyrophosphate to farnesol [60, 73, 86]. Like PAH1, DPP1 and LPP1 encode phosphatidate phosphatases. Dpp1 and Lpp1 only have minor effects on PL and TAG levels [87], but their deletion potentially influenced the growth and ABA titer of the pah1Δ strain yMO35.
Exchanging of the native PAH1 promoter to mediate growth deficiency
The deletion of PAH1 in yMO35 led to a severe growth defect, making the strain unsuitable for further engineering or future biotechnological uses. However, encouraged by the improved relative ABA titer of yMO35 (Fig. 4), we investigated if a gene knock-down could mediate the growth defect and still benefit ABA production. Expression levels of the PAH1 promoter during growth in glucose are comparable to the REV1 promoter and are <1% of the commonly used strong TEF1 promoter (unpublished data).
The native PAH1 promoter was exchanged for three different promoters to investigate their effects on growth and ABA production. We chose the constitutively weak promoter pREV1 (strain yMO36), the glucose-concentration-dependent promoter pHXT1 (strain yMO38) and a synthetic minimal promoter named pminCYC1 (strain yMO39) [88]. pHXT1 is repressed in low glucose conditions and is used in the background strain SCIGS22a to regulate ERG9 expression with the goal of separating cell growth and production phase [73, 89]. Different truncated versions of the CYC1 promoter have been used in other studies before and minimal expression levels were observed [88, 90]. In our study, we used a substantially truncated version (144 bp, sequence in Supplementary Table S6, Additional file 1), in which only one of three TATA elements remain [88, 91].
Figure 5A shows the OD_600_, absolute and relative ABA titers of strains with replaced pPAH1, the control strain with wild-type pPAH1 (yMO23) and the pah1Δ strain (yMO26). The strains with wild-type PAH1 and promoter replacement showed no significant difference in OD_600_. The pHXT1-PAH1 and pREV1-PAH1 modifications did not significantly improve ABA titers. However, the strain carrying the pminCYC1-PAH1 modification, yMO39, produced 15.8 mg/L ABA, ≈1.3-fold the titer produced by the wild-type PAH1 control yMO23. For all strains with modified PAH1 promoters, the ABA titer normalized to OD_600_ remained at PAH1 wild-type levels, which is about 40% lower than for the pah1Δ strain yMO26 (Fig. 5A).Fig. 5. Effects of PAH1 promoter replacement. The PAH1 promoter was exchanged for pREV1 (yMO36), pHXT1 (yMO38) or pminCYC1 (yMO39) and the effect was compared to the parent strain with wild-type pPAH1 (yMO23) or a pah1Δ strain (yMO26). Grey circles show the individual data points used to calculate the mean and standard deviation. Significance is shown as compact letter display above the bars. Shared letters indicate no significant difference. Wt wild-type. A OD_600_ (beige), ABA titer in the supernatant (green) and ABA titer normalized to OD_600_ (blue) are shown after 60 h of cultivation in mineral medium (24-deepwell microplates). B Growth profiles and maximum growth rate μ_max_ are shown for the background strain SCIGS22a (no B. cinerea genes, grey), the wild-type PAH1 control yMO23 (brown), the pah1Δ strain yMO26 (red) and yMO39 carrying the pminCYC1-PAH1 modification (dark blue). Lines in the growth profiles show the mean OD_600_ of three replicates and ribbons visualize their standard deviation. Cells were grown in mineral media (96-well microplates)
In addition, we compared the growth profile and maximum growth rate μ_max_ of the background strain SCIGS22a, the wild-type PAH1 strain yMO23, the pah1Δ strain yMO26 and the pminCYC1-PAH1 strain yMO39 (Fig. 5B). The growth profiles and μ_max_ of yMO23 and yMO39 were similar, both had a lower μ_max_ and slightly lower final OD_600_ than the background strain. Among the analysed strains, the pah1Δ strain yMO26 had the most severe growth defect in terms of μ_max_ and final OD_600_. Interestingly, yMO26 did not show bi-phasic growth. Deletion of PAH1 has been reported to cause respiratory deficiencies [92], potentially explaining the growth defect.
In conclusion, by exchanging pPAH1 for the weak minimal promoter pminCYC1, we were able to avoid the growth defect observed in the pah1Δ strain yMO26, while still improving ABA titers. The relative ABA titer per OD_600_ was higher for yMO26 than for yMO39, indicating that ABA production might be further improved by fine-tuning ER proliferation. We assume that pPAH1 replacement could be a promising engineering strategy in Y. lipolytica ABA cell factories.
Expression of A. thaliana proteins to improve ABA production
The A. thaliana proteins AtMSBP1, AtCOL4 and AtGRP7 were previously found to significantly improve CYP activity in a betaxanthin-producing S. cerevisiae strain [39]. Motivated by this, we sought to investigate the proteins’ effects on ABA production. However, difficulties were encountered during the chemical synthesis of the AtGRP7 gene. AtGPR7 also only had minor effects on CYP activity in previous work [39] and we therefore decided to exclude the protein from our study. AtMSBP1 and AtCOL4 were individually expressed by integrating a single copy of each gene into the ySMC001 genome (a strain based on SABA3 with single copies of each bcaba gene and bccpr1), controlled by strong constitutive promoters pTEF2 for AtMSBP1 and pHHF2 for AtCOL4, resulting in the strains ySMC023 and ySMC024, respectively. The strain overexpressing AtMSBP1 produced 16.6 mg/L ABA, a 3.5-fold increased ABA titer compared to ySMC001 (Fig. 6). While expression of AtCOL4 alone did not influence ABA production, co-expression of both, AtMSBP1 and AtCOL4, in strain ySMC025 significantly increased the titer to 26 mg/L, a 5.5-fold increase compared to the starting strain ySMC001 and a 1.6-fold increase to ySMC023 carrying solely AtMSBP1. The relative ABA titer for ySMC023 and ySMC025 was also improved compared to ySMC001, however no significant difference between ySMC023 and ySMC025 was observed (Fig. 6). Expression of the A. thaliana genes did not affect OD_600_.Fig. 6. Effects of AtMSBP1 and AtCOL4 expression and ERG20 overexpression. The Arabidopsis genes were integrated in the ySCM001 background (single copies of bcaba1/2/3/4 and bccpr1) or yMO39 background (two copies of bcaba1/2, expression of bccyb5 and bccbr1 as well as the pminCYC1-PAH1 modification). An additional copy of ERG20 was added to improve precursor supply. “X” indicates presence of the genetic modification in the strain. OD_600_ (beige), ABA titer in the supernatant (green) and ABA titer normalized to OD_600_ (blue) are shown after 48 h of cultivation in mineral media (24-deepwell microplates). Grey circles show the individual data points used to calculate the mean and standard deviation. Significance is shown as compact letter display above the bars. Shared letters indicate no significant difference
To further improve ABA production, we added the AtMSBP1 cassette alone or in combination with AtCOL4 to the previously best performing strain yMO39, resulting in strains ySMC027 and ySMC028. Besides the A. thaliana genes, these strains also carry additional copies of bcaba1 and bcaba2, express bccyb5 and bccbr1 and include the pPAH1Δ::pminCYC1 modification. Expression of AtMSBP1 in strain ySMC027 did not alter OD_600_, nor did it affect absolute or normalized ABA titer compared to yMO39 (Fig. 6). Co-expression of AtMSBP1 and AtCOL4 in ySMC028 severely reduced OD_600_ while no beneficial effect was visible for the absolute ABA titer. However, when normalized to the OD_600_, the ABA titer was increased in this strain.
Finally, we investigated whether further enhancing the MVA pathway flux could benefit ABA production. To achieve this, we expressed an additional copy of ERG20 under the control of pTEF2 in the strains ySMC025 and ySMC028 resulting in strains ySMC026 and ySMC029, respectively. No significant increase in absolute ABA titers was achieved when compared to the respective parental strains (Fig. 6). While the addition of an extra ERG20 gene in the strain ySMC026 did not result in a higher ABA/OD_600_ ratio, in the ySMC029 strain, the additional copy resulted in the highest ABA/OD_600_ ratio of 9.3 (Fig. 6). These results support the idea that adding an extra copy of ERG20 directs the pathway flux toward product formation. The effect is particularly important in strains derived from the yMO39 background; however, the OD_600_ was severely impaired in this strain, likely due to the metabolic burden caused by extensive genetic engineering.
Strikingly, the titers of ABA achieved by the highly engineered strain ySMC027 (21.90 mg/L) were comparable to those obtained in the less modified strain ySMC025 (26.22 mg/L), in which only a single copy of the P450s, bcaba1 and bcaba2, are present (Fig. 6). The expression of the Arabidopsis genes seems to be the most straightforward and effective strategy, as it requires few engineering steps and does not adversely affect the OD_600_.
We speculate that the enhancement in ABA production observed with At**MSBP1 expression is likely due to its dual role as a chaperone-like protein and a structural scaffold for the assembly of ER-bound enzymes [40, 93–95]. Subcellular localization studies showed that MSBP1 from Saponaria vaccaria co-localizes with CYPs on the ER membrane of yeast [93, 94], suggesting a potential interaction that could be leveraged for enhanced ABA biosynthesis. The boost in ABA production observed when AtCOL4 is co-expressed with AtMSBP1, but not for AtCOL4 alone, suggests that the two Arabidopsis proteins interact. While AtCOL4 is known to enhance abiotic stress tolerance and regulate ABA biosynthesis genes in Arabidopsis [96], its molecular mechanisms in yeast remains unclear. It is worth noting that the ABA biosynthetic pathway differs substantially in plants and fungi [10] and the effect of AtCOL4 is not specific for enzymes involved in ABA production [39]. In ySMC027 the higher expression levels of bcaba1 and bcaba2, the presence of bccyb5 and bccbr1, and the *PAH1-*modulation-mediated ER alterations might result in non-optimal assembly of enzyme complexes (i.e. due to different stoichiometries), potentially interfering with the AtMSBP1- and AtCOL4-mediated effect. Further studies will be required to elucidate the underlying mechanism.
The hypothesis that enzyme complexes on the ER-membrane can improve enzyme activity is also supported by a recent study by Sun et al. [20]. The authors found that linking BcABA1 and BcABA2 via short peptide tags improved ABA production in Y. lipolytica by 38%. This beneficial effect appears to be enzyme-specific since linking BcABA3 and an ERG20 mutant with the same peptide tags did not increase ABA titers [93].
During the revision of this paper, a new study reported that expression of AtMSBP1 broadly enhanced plant CYP performance in yeast [97], providing an additional example of its effectiveness. Importantly, the authors observed that AtMSBP1 expression induces not only ER expansion but also causes an increase in mitochondrial volume and inducing vacuole fission [97] . Furthermore, overexpression of the genes encoding proteins involved in organelle coordination such as RTN1 (reticulon 1), VMA2 (vacuolar ATPase subunit), AFG1 (ATPase family gene 1), MERG3 (mitochondrial genome regulator) and PHB1 (prohibitin 1) enhanced CYP efficiency*.* Interestingly, co-expressing AtMSBP1 with these proteins did not result in an additive effect on 1-OH-N-methylcanadine production, suggesting redundancy in their roles as CYP-supporting proteins. This is similar to the apparent redundancy we observed when combining AtMSBP1 expression with the PAH1 knockdown. Nonetheless, our findings open new avenues for designing and fine-tuning strategies that integrate AtMSBP1 and AtCOL4 expression with additional organelle-remodeling targets to optimize yeast cells factories for ABA production.
Conclusion
The microbial production of ABA through engineered cell factories offers a promising and sustainable alternative to other methods such as extraction from plants or chemical synthesis. While the ABA biosynthetic pathway can be successfully reconstituted in S. cerevisiae, optimizing its performance remains challenging, particularly due to the complexity of CYP enzyme activity in relation to the cellular environment. We investigated various strategies to enhance ABA production in yeast, including modulation of transporter expression, adding additional CYP gene copies, expressing CYB5, improving heme and oxygen supply, ER proliferation and heterologous expression of A. thaliana genes. Surprisingly, we found that co-expression of the Arabidopsis genes AtMSBP1 and AtCOL4 improved ABA titers several fold. Combining the most promising engineering strategies, namely additional CYP gene copies, ER proliferation and Arabidopsis gene expression did not result in further improvements. This and other recent studies show that optimizing CYP functionality in cell factories is complex and its underlying principles are still not completely understood. In this regard, our findings provide valuable insights not just for heterologous ABA biosynthesis but also for potential CYP-expression platform strains.
Supplementary Information
Additional file 1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Finkelstein R. Abscisic acid synthesis and response. The Arabidopsis Book. Vol 11. 2013.10.1199/tab.0166 PMC 383320024273463 · doi ↗ · pubmed ↗
- 2Ortiz-García P, González Ortega-Villaizán A, Onejeme FC, et al. Do opposites attract? Auxin-abscisic acid crosstalk: new perspectives. Int J Mol Sci. 2023;24:3090. 10.3390/ijms 24043090 PMC 996082636834499 · doi ↗ · pubmed ↗
- 3Gupta K, Wani SH, Razzaq A, et al. Abscisic acid: role in fruit development and ripening. Front Plant Sci. 2022;10;13:817500. 10.3389/fpls.2022.817500 PMC 912766835620694 · doi ↗ · pubmed ↗
- 4Olds CL, Glennon E, Luckhart S. Abscisic acid: new perspectives on an ancient universal stress signaling molecule. Microbes Infect. 2018;20:484–92. 10.1016/j.micinf.2018.01.00929408537 · doi ↗ · pubmed ↗
- 5Kim SW, Goossens A, Libert C, et al. Phytohormones: multifunctional nutraceuticals against metabolic syndrome and comorbid diseases. Biochem Pharmacol. 2020;175:113866.10.1016/j.bcp.2020.11386632088261 · doi ↗ · pubmed ↗
- 6Shi T-Q, Peng H, Zeng S-Y, et al. Microbial production of plant hormones: opportunities and challenges. Bioengineered. 2016;8:124–8. 10.1080/21655979.2016.1212138 PMC 539860227459344 · doi ↗ · pubmed ↗
- 7Siewers V, Smedsgaard J, Tudzynski P. The P 450 monooxygenase Bc ABA 1 is essential for abscisic acid biosynthesis in Botrytis cinerea. Appl Environ Microbiol. 2004;70. 10.1128/AEM.70.7.3868-3876.2004 PMC 44475515240257 · doi ↗ · pubmed ↗
- 8Jiang L, Dong C, Liu T, et al. Improved functional expression of cytochrome P 450s in Saccharomyces cerevisiae through screening a c DNA library from Arabidopsis thaliana. Front Bioeng Biotechnol. 2021;9:764851. 10.3389/fbioe.2021.764851 PMC 869602734957066 · doi ↗ · pubmed ↗