A Rare Case of Coexisting Medullary and Papillary Thyroid Carcinomas With Parathyroid Adenoma in Pancreatic Neuroendocrine Tumour (pNET)
Nazia Binte Salam, Fahmida Tithi, Sunil Zachariah

TL;DR
A 79-year-old man had rare coexisting thyroid and parathyroid tumors along with a pancreatic tumor, highlighting the need for multidisciplinary care.
Contribution
This case report documents the rare coexistence of multiple endocrine tumors and suggests potential syndromic associations.
Findings
The patient had medullary and papillary thyroid carcinomas along with a parathyroid adenoma.
An incidental pancreatic neuroendocrine tumor was also identified.
The case emphasizes the importance of multidisciplinary collaboration for complex diagnoses.
Abstract
We present a rare and complex case involving the simultaneous occurrence of medullary thyroid carcinoma (MTC), papillary thyroid carcinoma (PTC), and a parathyroid adenoma in a 79-year-old male. Additionally, incidental radiological findings suggest a pancreatic neuroendocrine tumour (NET). The coexistence of these diverse endocrine disorders is exceptionally uncommon and raises important questions regarding potential syndromic associations. Such cases highlight the necessity for thorough diagnostic evaluation, individualised treatment planning, and the collaboration of a multidisciplinary team, including endocrinologists, oncologists, surgeons, radiologists, and pathologists, to ensure optimal patient outcomes.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
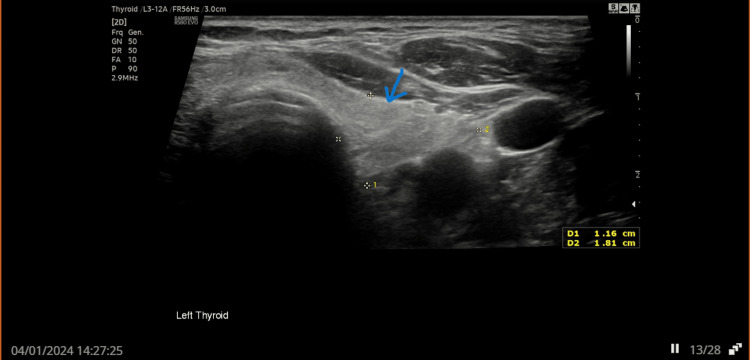 Figure 1
Figure 1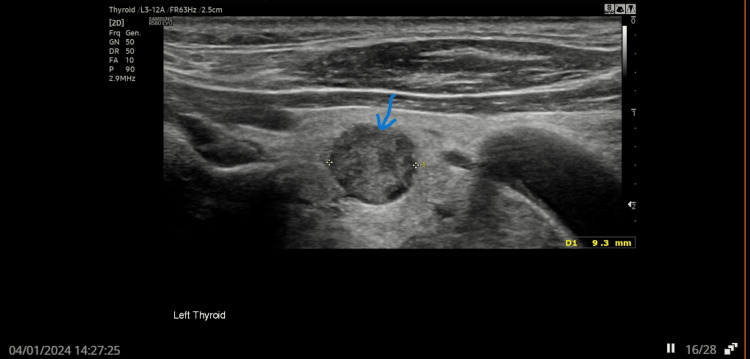 Figure 2
Figure 2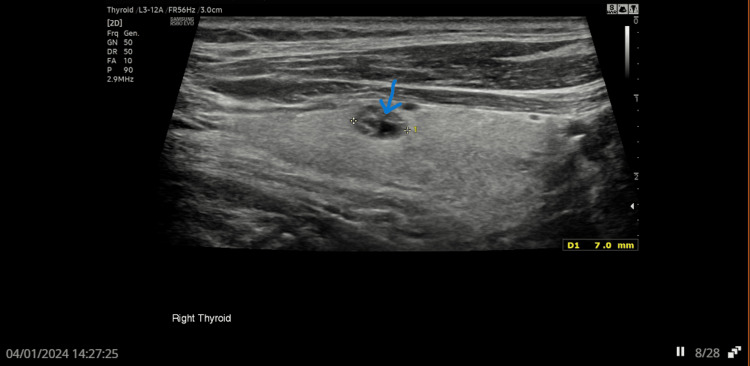 Figure 3
Figure 3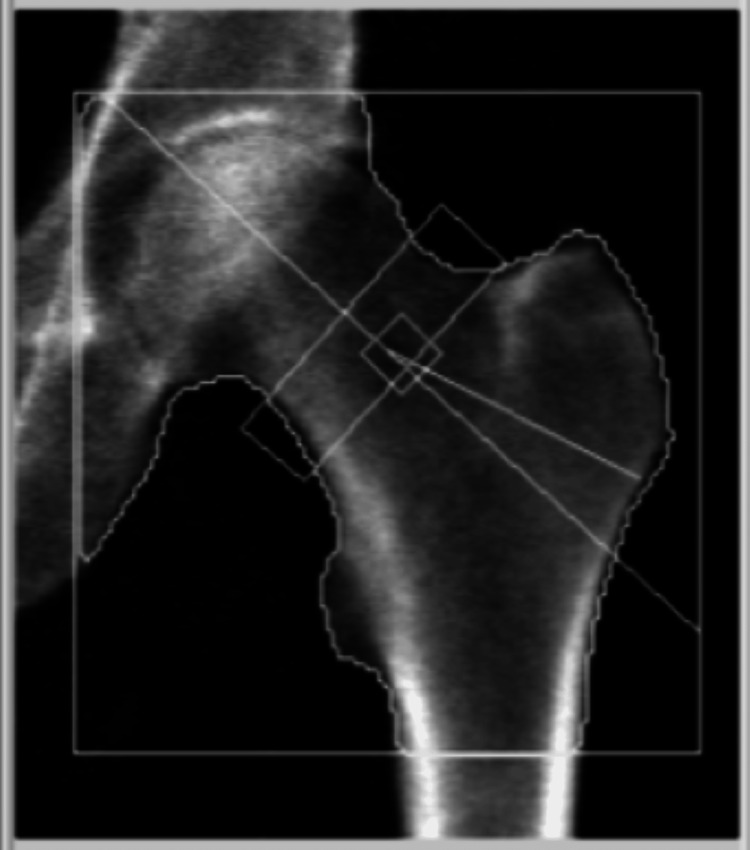 Figure 4
Figure 4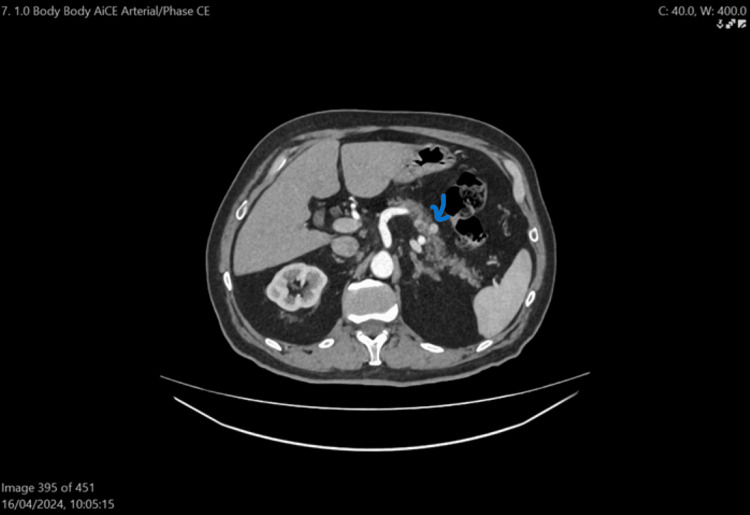 Figure 5
Figure 5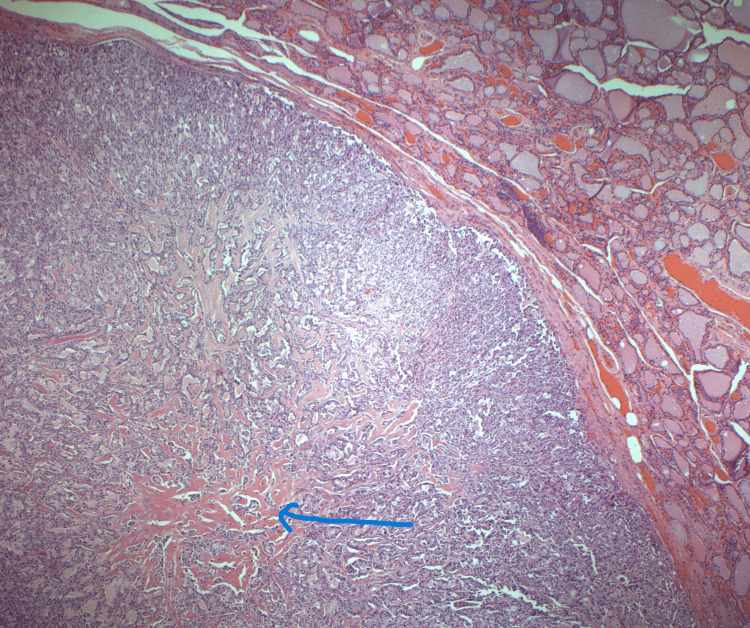 Figure 6
Figure 6| Biochemistry | Normal range | April 2023 | September 2023 | January 2024 | March 2024 | June 2024 | July 2024 |
| Parathyroid | 1.6- 7.2 pmol/L | 21.5 | N/A | 15.6 | 13.4 | N/A | N/A |
| Serum Adjusted Calcium | 2.2- 2.6 mmol/L | 2.44 | 2.45 | 2.54 | 2.56 | N/A | 2.31 |
| Serum Phosphate | 0.8-1.5 mmol/L | 0.65 | 0.75 | 0.64 | 0.80 | N/A | N/A |
| Serum Carcinoembryonic antigen | 0-4.9 ug/L | N/A | N/A | N/A | 8.8 | N/A | <1.7 |
| Serum Calcitonin | 0-11.8 ng/L | N/A | N/A | N/A | 164 | N/A | 1.2 |
| Vasoactive intestinal peptide | 0-30 pmol/L | N/A | N/A | N/A | N/A | 15 | N/A |
| Pancreatic polypeptide | 0-300 pmol/L | N/A | N/A | N/A | N/A | 54 | N/A |
| Gastrin | 0-48 pmol/L | N/A | N/A | N/A | N/A | 168 | N/A |
| Glucagon | 0-50 pmol/L | N/A | N/A | N/A | N/A | 72 | N/A |
| CART | 0-129 pmol/L | N/A | N/A | N/A | N/A | 38 | N/A |
| Somatostatin | 0-150 pmol/L | N/A | N/A | N/A | N/A | 44 | N/A |
| Chromogranin A | 0-60 pmol/L | N/A | N/A | N/A | N/A | 17 | N/A |
| Chromogranin B | 0-150 pmol/L | N/A | N/A | N/A | N/A | 66 | N/A |
| Urine non metadrenaline | 0-3.3 umol/24h | N/A | N/A | N/A | 2.02 | N/A | N/A |
| Urine Metadrenaline | 0-1.2 umol/24h | N/A | N/A | N/A | 0.68 | N/A | N/A |
| Urine 3 Methoxytyramine | 0-2.5 0.68 umol/24h | N/A | N/A | N/A | 0.68 | N/A | N/A |
| Region | Area(cm2) | BMC[(g)] | BMD[g/cm2] | T-score | PR (Peak Reference) | Z-score | AM (Age Matched) |
| Neck | 6.21 | 4.08 | 0.657 | -2.0 | 71 | -0.6 | 89 |
| Total | 45.04 | 39.39 | 0.875 | -1.0 | 85 | -0.1 | 98 |
| 10-year Fracture Risk | |
| Major Osteoporotic Fracture | 6.5% |
| Hip Fracture | 2.5% |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsThyroid Cancer Diagnosis and Treatment · Neuroendocrine Tumor Research Advances · Thyroid and Parathyroid Surgery
Introduction
Pancreatic neuroendocrine tumours (pNETs) are uncommon malignancies, representing only 1-2% of all pancreatic neoplasms. Marini et al. found 10% of pNETs occur in the context of hereditary endocrine syndromes, notably multiple endocrine neoplasia type 1 (MEN1) [1]. The estimated annual incidence is one to two cases per 100,000 individuals. Kamei et al. mentioned that most pNETs are well-differentiated neoplasms that tend to grow slowly [2]. Li et al. classified pNETs into functional tumours, which secrete hormones that induce characteristic clinical syndromes, and nonfunctional tumours, which often lack overt hormonal manifestations [3]. Kamei et al. reported that early symptoms are usually vague or absent, that diagnosis is frequently delayed, and that many patients present with advanced disease [2].
McDonnell et al. mentioned MEN1 includes pancreatic tumours, pituitary tumours, and primary hyperparathyroidism. MEN2 (also known as MEN2A) consists of primary hyperparathyroidism, pheochromocytoma, and medullary thyroid cancer [4]. Additionally, Marfanoid habitus, pheochromocytoma, medullary thyroid cancer, and neuroma are seen in MEN3 (also known as MEN2B). Notably, there is an overlap between certain conditions and the MEN association. McDonnell et al. emphasized that a high index of diagnostic suspicion is required because of variable presentations of these conditions, and suspected patients should be promptly referred to a specialized centre for early initiation of treatment and regular follow-up [4]. MEN2, MEN3 and caused by mutations in the rearranged during transfection (RET) proto-oncogene. McDonnell et al. also stated that there is an established genotype-phenotype association in MEN2 and MEN3, but not in MEN1 [4].
McDonnell et al. mentioned that treatment depends on the cause. Surgical resection is the definitive treatment for parathyroid, pancreatic, and thyroid tumours [4]. Recently, medullary thyroid carcinoma (MTC) has been successfully treated with tyrosine kinase inhibitors. Vandetanib, an oral inhibitor of RET, vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor signalling, has shown long-lasting disease control in patients with advanced hereditary MTC.
Remarkably, about 95% of thyroid malignancies are differentiated thyroid carcinomas (DTCs), most commonly papillary thyroid carcinoma (PTC), which arises from follicular cells and accounts for nearly 85% of cases. Fallahi et al. found MTC arises from parafollicular C cells and is a distinct entity, with roughly 75% of cases occurring sporadically and 25% in hereditary forms, often linked to MEN types 2A and 2B [5]. Fallahi et al. also mentioned that the concurrent occurrence of PTC and MTC is rare [5]. Even more uncommon is the simultaneous presentation of MTC, PTC, parathyroid adenoma, and a suspected pancreatic NET.
Case presentation
A 79-year-old man was referred to the endocrine team in early 2023 by his GP due to persistently elevated parathyroid hormone (PTH) levels, despite normal serum calcium and low phosphate. Initial thyroid ultrasound in June 2023 revealed an isoechoic U2 nodule in the left lobe (Figure 1) and hypoechoic U3 nodules in both lobes of the thyroid (Figures 2, 3). Follow-up imaging in January 2024 showed no significant change.
Ultrasound thyroid showing isoechoic U2 nodule in the left lobeArrow showing isoechoic U2 nodule in left lobe of thyroid.U2: benign or non-cancerous nodule.
US thyroid showing hypoechoic U3 nodule in the left lobeArrow showing hypoechoic U3 nodule in left lobe of thyroid.U3: equivocal nodule. The suspicion of malignancy is low to moderate.
US thyroid showing hypoechoic nodule in right lobeArrow showing hypoechoic nodule in right lobe of thyroid.
He was asymptomatic, with no palpable neck mass or signs of compression, but was referred to Ear, Nose, and Throat (ENT) team via the two-week cancer pathway. Thyroid function tests, autoantibodies, calcium, vitamin D, and PTH levels were reassessed. Fine-needle aspiration cytology (FNAC) of the left U3 thyroid nodule in February 2024 showed features suspicious for medullary thyroid carcinoma or Hurthle cell lesion. Further blood tests revealed elevated calcitonin, carcinoembryonic antigen (CEA), persistently elevated PTH, and calcium (Table 1). Urinary metanephrines were negative. A dual X-ray absorptiometry (DEXA) scan in April 2024 revealed osteopenia (Figure 4, Tables 2, 3).
Dual X-ray absorptiometry (DEXA) scan image
Tables 2, 3 showed osteopenia, prompting further assessment and an endocrine MDT discussion.
In 2024, a contrast-enhanced CT scan of the neck and thorax abdomen revealed no evidence of an aerodigestive mass. Previously known thyroid nodules were noted, along with mildly prominent lymph nodes at levels 3 and 4 on the left. A 9 mm suspected parathyroid lesion was identified along the left lateral wall of the oesophagus at the C7-T1 level. Additionally, a calcified nodule was observed in the right upper lobe of the lung, but no suspicious pulmonary lesions or lymphadenopathy were present. An 11 mm enhancing nodule in the pancreatic body (Figure 5) raised concern for malignancy, and a gastrointestinal review was recommended. Gallstones were also detected. There was no pleural effusion, pulmonary embolism, breast mass, or aggressive bone lesions.
CT Neck Thorax Abdomen showing pancreatic noduleArrow showing pancreatic nodule.
The thyroid MDT in April 2024 recommended further assessment of the pancreatic lesion. An MRI pancreas showed an 8 mm lesion in the tail with a differential diagnosis of intrapancreatic accessory spleen versus NET. A PET dotatate scan in May 2024 confirmed two dotatate-avid pancreatic foci, indicative of well-differentiated NETs.
The patient underwent total thyroidectomy with parathyroidectomy on 30th May 2024. Histological examination confirmed a 9 mm medullary thyroid carcinoma (Figure 6) in the left lobe (pT1aN0M0) with focal amyloid deposition. In addition, incidental bilateral papillary thyroid carcinomas (pT1aN0M0) were identified. A benign parathyroid adenoma was also noted. All 28 lymph nodes examined were negative for malignancy.
Histology image showing medullary thyroid carcinoma
Postoperatively, the patient showed normalized biochemical markers: calcium, calcitonin, and CEA as of July 2024 (Table 1). Genetic testing in September 2024 was negative for RET mutations or MEN syndromes.
Discussion
This case underscores a highly unusual confluence of endocrine pathologies. MTC accounts for only 3-4% of all thyroid cancers, and its coexistence with PTC - arising from entirely different cell lines - is rare, with few cases documented. Even rarer is the concurrent presence of a parathyroid adenoma and a suspected pancreatic NET, raising suspicion of MEN2, although genetic testing was negative.
The patient’s mildly elevated calcium, high PTH, and the CT finding of a lesion adjacent to the oesophagus were consistent with a parathyroid adenoma. Notably, his thyroid ultrasound, initially performed for parathyroid localisation, incidentally identified the U3 nodule that ultimately revealed MTC. This signifies the diagnostic value of incidental findings and broad-spectrum imaging.
High calcitonin level in this case favoured the diagnosis of MTC. Raised CEA also supported pancreatic and thyroid carcinoma. Parathyroid hormone level was three times the upper limit of normal, which came down after treatment. In summary, a combination of elevated calcitonin levels, Ultrasound findings, FNAC, and PET uptake supports the diagnosis of MTC and prompts treatment. PET dotatate avidity in the pancreas suggests the possibility of synchronous NETs. While histological confirmation was not yet available at the time of this report, the pancreatic lesions remain under surveillance by the upper gastrointestinal (GI) team.
Multidisciplinary collaboration between endocrinology, radiology, ENT, surgery, and upper GI specialists enabled timely and effective diagnosis and management. Importantly, the absence of genetic mutations indicates that this complex presentation may be sporadic rather than syndromic.
Conclusions
This case illustrates the diagnostic challenges posed by multiple endocrine abnormalities. Incidental imaging of a thyroid nodule and biochemical screening revealed the syndromic-like presentations. This also reinforces that negative genetic testing does not exclude complex sporadic endocrine tumour combinations. Clinicians should be more aware of concurrent endocrine pathology when encountering biochemical-radiological discrepancies. The rare synchronous occurrence of MTC, PTC, a parathyroid adenoma, and a probable pancreatic NET underscores the importance of comprehensive evaluation and multidisciplinary collaboration. Despite the absence of a known hereditary syndrome, continued follow-up and surveillance are crucial given the potential for recurrence or additional endocrine pathology.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pancreatic neuroendocrine neoplasms in multiple endocrine neoplasia type 1Int J Mol Sci Marini F Giusti F Tonelli F 222021 https://doi.org/10.3390/ijms 2208404110.3390/ijms 22084041 PMC 807078833919851 · doi ↗ · pubmed ↗
- 2Neuroendocrine tumor of the small intestine: case report Arq Bras Cir Dig Kamei DJ Shiguihara RS Araújo FR 0332020 https://doi.org/10.1590/0102-672020190001 e 149210.1590/0102-672020190001 e 1492 PMC 723632732428136 · doi ↗ · pubmed ↗
- 3Pancreatic neuroendocrine tumor producing vasopressin: a case report Medicine (Baltimore) Li J Zhang X He Q 01002021 http://dx.doi.org/10.1097/MD.000000000002745310.1097/MD.0000000000027453 PMC 850060734622867 · doi ↗ · pubmed ↗
- 4Multiple endocrine neoplasia: an update Intern Med J Mc Donnell JE Gild ML Clifton-Bligh RJ 954961492019 https://doi.org/10.1111/imj.143943138715610.1111/imj.14394 · doi ↗ · pubmed ↗
- 5Simultaneous occurrence of medullary thyroid carcinoma and papillary thyroid carcinoma: a case series with literature review Curr Oncol Fallahi P Patrizio A Stoppini G 1023710248302023 https://doi.org/10.3390/curroncol 301207453813237910.3390/curroncol 30120745 PMC 10742226 · doi ↗ · pubmed ↗
- 6Clinical complexity in osteopenia BMJ Pilz S Schmitt L Kraus DA 3912025 https://doi.org/10.1136/bmj.r 2154 10.1136/bmj.r 215441125421 · doi ↗ · pubmed ↗
- 7The Fracture Risk Assessment Tool (FRAX®): applications in clinical practice J Womens Health Watts NB 202011 https://doi.org/10.1089/jwh.2010.229410.1089/jwh.2010.229421438699 · doi ↗ · pubmed ↗