PPE64 is a mycomembrane channel protein that functions in heme iron uptake and moonlights in biofilm formation in Mycobacterium tuberculosis
Padam Singh, Charles B. Kaufman, Lisa Whitworth, Reed M. Stubbendieck, Randy Morgenstein, Karen L. Wozniak, Avishek Mitra

TL;DR
This study shows that the PPE64 protein in tuberculosis bacteria helps the bacteria take in iron from the host and also helps them form biofilms, which are important for their survival.
Contribution
The study reveals PPE64's dual role in heme iron uptake and biofilm formation, with distinct oligomeric states for each function.
Findings
PPE64 is essential for heme iron acquisition in Mycobacterium tuberculosis.
PPE64 contributes to biofilm formation independently of iron availability.
PPE64's absence impairs Mtb growth in human macrophages.
Abstract
Mycobacterium tuberculosis (Mtb) is the leading cause of human deaths by an infectious agent. To survive in the human host, Mtb must acquire essential iron nutrients from the host and evade the immune response. In diderm bacteria like Mtb, outer membrane channel transporter proteins are fundamental for nutrient acquisition and immune evasion. Recently, we demonstrated that the Mtb outer mycomembrane PPE64 is a channel protein, providing the first direct evidence of channel-forming capability by a protein of the PPE (proline-proline-glutamate motif) family, which are found exclusively in mycobacteria. Here, we demonstrate that the PPE64 channel protein is specifically required for the uptake of heme, which is the largest source of iron in the human host. Furthermore, PPE64 plays a crucial role in biofilm formation, and this function is not dependent on the iron source in the medium. The…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
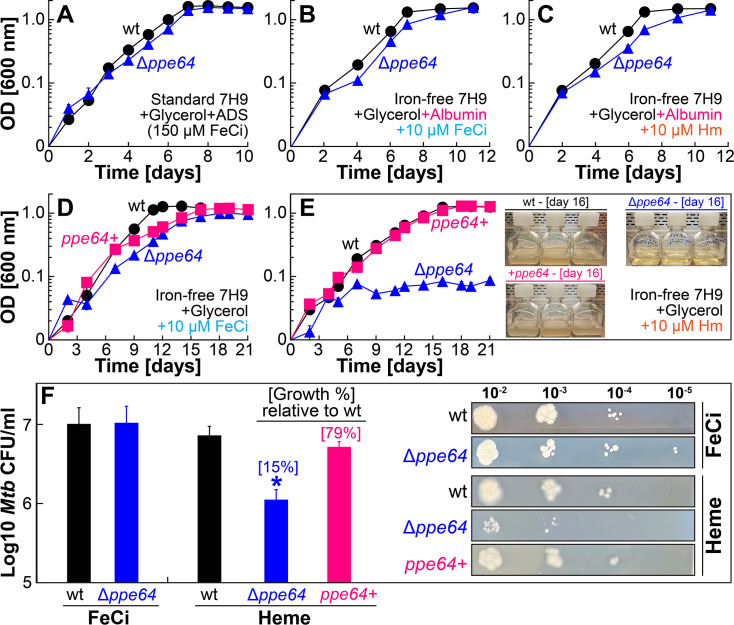 Fig 1
Fig 1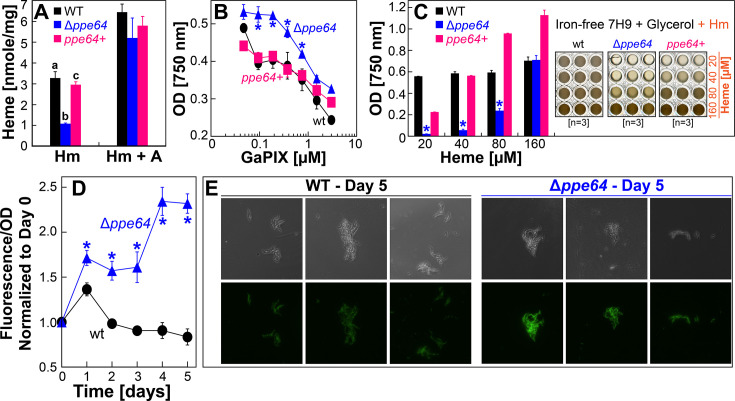 Fig 2
Fig 2 Fig 3
Fig 3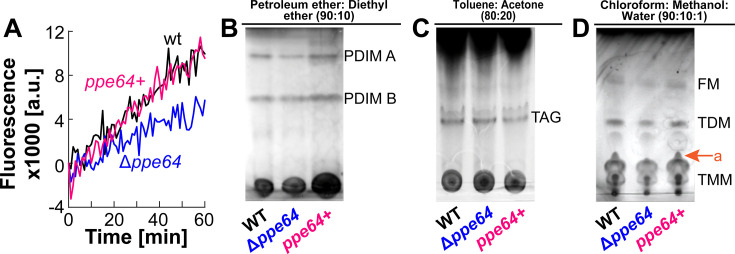 Fig 4
Fig 4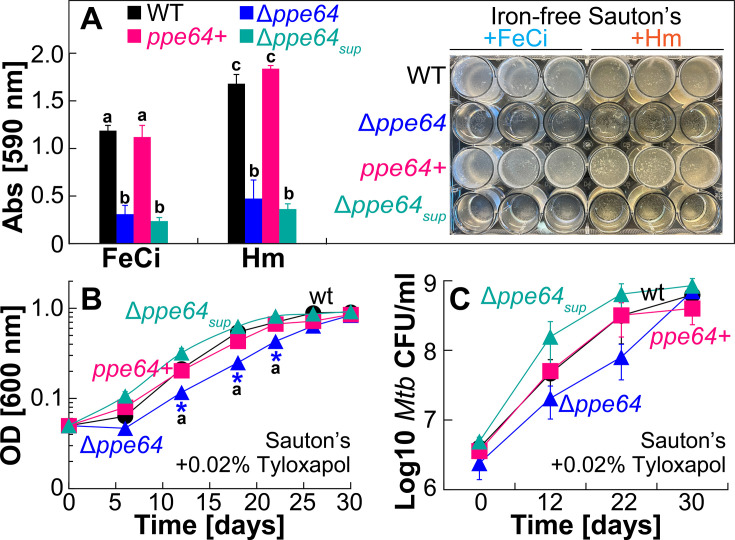 Fig 5
Fig 5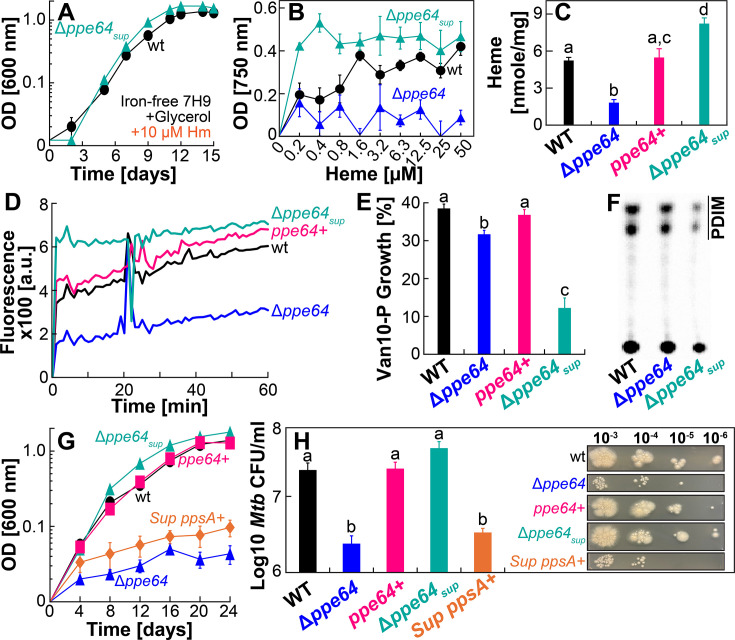 Fig 6
Fig 6 Fig 7
Fig 7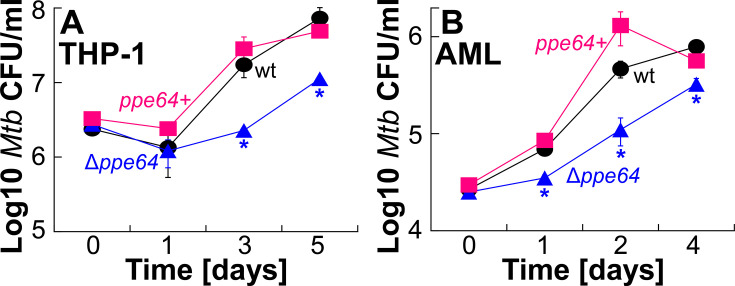 Fig 8
Fig 8- —National Institute of General Medical Scienceshttp://dx.doi.org/10.13039/100000057
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTuberculosis Research and Epidemiology · Bacterial Genetics and Biotechnology · Bacterial biofilms and quorum sensing
INTRODUCTION
Mycobacterium tuberculosis (Mtb) kills ~1.3 million people yearly and has surpassed HIV/AIDS to become the leading cause of human deaths by an infectious agent (1). The ability of Mtb to overcome nutrient limitations in the host and evade the immune response is the principal reason that makes it such a successful pathogen. Following Mtb entry into the human lung, resident macrophages phagocytose Mtb and activate the nutritional immunity response to sequester metals (2, 3) like iron, zinc, manganese, and magnesium, which are required for vital biological functions (4). In this intracellular environment, Mtb is constantly battling to acquire metal nutrients from the host for its own survival. However, Mtb also spends a key part of its life cycle as extracellular bacilli within the granuloma (5). Numerous studies have shown that extracellular Mtb grow within biofilms and have linked biofilm growth to immune evasion and antibiotic treatment tolerance (6–10). These observations are not surprising given that bacteria primarily exist within biofilms in nature and bacterial biofilms are major drivers of chronic infections and antibiotic tolerance (11–13). Thus, understanding the principles of Mtb nutrient acquisition and biofilm formation is of great relevance, as this knowledge will allow us to develop targeted strategies to inhibit vital processes in Mtb.
Diderm bacteria (like Mtb [14, 15]) use canonical β-barrel outer membrane proteins (OMPs) to traffic molecules across their outer membrane. Expectedly, β-barrel OMPs are central to the survival and pathogenicity of gram-negative bacteria because they function in fundamental biological processes, such as uptake of essential metal nutrients (2, 16), outer membrane stabilization (17), motility (18), biofilm formation (19–21), and antibiotic resistance (22). Many studies (23, 24) have employed bioinformatic approaches to identify putative Mtb β-barrel OMPs and some Mtb mycomembrane proteins (25–28) with channel-forming capability and solute transport have been identified. However, we still do not have direct structural evidence of Mtb β-barrel OMPs. In recent years, a growing body of evidence has shown that some proteins of the proline-proline-glutamate (PPE) motif family function in transporting molecules across the Mtb mycomembrane. For example, PPE36/PPE62 (29, 30), PPE37 (31), and PPE4 (32) function in iron acquisition, PPE20 (33) and PPE31 (34) function in calcium and magnesium transport, respectively, and PPE51 (34–38) functions in the trafficking of various nutrient molecules. Notably, PPE proteins are exclusively found in mycobacteria (39, 40). These observations led to a hypothesis that some proteins of the PPE family can function as channel proteins to transport molecules across the mycomembrane.
In our most recent study, we provided the first direct evidence for this emerging hypothesis by demonstrating that PPE64 is a mycomembrane protein that forms channels in lipid membranes (41). In our current study, we demonstrate that the PPE64 channel protein is essential for heme (Hm) iron uptake. This is of great significance because the majority (>75%) of the iron in our body is stored as Hm within hemoglobin (42) in blood, making Hm the largest reservoir of circulating iron in the human host (43, 44). Thus, PPE64 plays an important role in Mtb iron acquisition. PPE64 also has broader roles in Mtb physiology influencing cell size and biofilm formation. Finally, we demonstrate that these distinct roles of PPE64 are necessary for Mtb growth within human macrophages, establishing PPE64 as an important factor for Mtb virulence.
MATERIALS AND METHODS
Bacterial strains, growth media, and preparation of iron-free medium
Wild-type (wt) Mtb H37Rv and its derivative strains were grown in Middlebrook liquid 7H9 or solid 7H10 medium supplemented with 10% ADS (8.5 g/L NaCl, 20 g/L dextrose, and 50 g/L bovine albumin fraction V), 0.5% glycerol, 0.2% casamino acids (CAAs), and 0.02% tyloxapol. This fully supplemented medium is referred to as complete 7H9 medium from hereon. Biofilm assays for Mtb strains were performed in standard Sauton’s medium containing 0.5 g/L K_2_HPO_4_, 0.5 g/L MgSO_4_, 4.0 g/L L-asparagine, 2 g/L citric acid, 6% glycerol, and 0.05 g/L ferric ammonium citrate with a final pH of 7.2. Escherichia coli DH5α was grown in either LB medium containing appropriate antibiotics at 37°C with shaking at 180 rpm. The following antibiotics were used when required: ampicillin (Amp) at 100 μg/mL for E. coli; kanamycin (Kan) at 30 μg/mL for mycobacteria and 50 μg/mL for E. coli; and hygromycin (Hyg) at 200 μg/mL for E. coli and 50 μg/mL for mycobacteria.
Both 7H9 and Sauton’s contain ~150 µM ferric citrate. For iron-free 7H9 or iron-free Sauton’s, all components except ferric citrate were dissolved in Millipore water in acid-washed beakers to prepare the base iron-free media. Lyophilized albumin was added to iron-free 7H9 base medium to prepare the base iron-free albumin-7H9 medium. Medium was then filter sterilized through a 0.2 μM filter. Freshly made ferric citrate or hemin solution was added to either base medium to prepare the specific iron-containing medium. Hemin solutions at 20 mM concentration were prepared in DMSO. The iron chelators 2,2-dipyridyl (DIP) and deferoxamine (DFO) were prepared in DMSO and added to freshly made hemin medium.
Targeted gene deletion in Mtb and construction of gene expression vectors
To construct mutants, 1,000 bp of left (L) and 1,000 bp right (R) flanking sequences of the target gene were amplified using corresponding primer pairs LF/SpeI-LR/SwaI and RF/PacI-RR/NsiI (Table S3), respectively, and cloned into pML2424 to construct gene deletion vectors (Table S2). The deletion vectors were then transformed into Mtb. Transformants were selected at 37°C on 7H10 Hyg and visually validated through the presence of both GFP and RFP fluorescence. Liquid culture of transformant was then plated on 7H10 Hyg containing 2% sucrose at 40°C for selection of double crossovers. Putative double crossovers were visually analyzed for the presence of only GFP, and gene deletion was validated by PCR. To excise the loxP-flanked gfp^2+^m-hyg cassette, pML2714-expressing Cre recombinase was transformed into marked mutants, and unmarked mutants were selected on 7H10 Kan at 37°C. Putative unmarked mutants were first visually validated through the absence of GFP fluorescence and then through PCR (Fig. S1) (validation primers (V/F – V/R), Table S3) and loss of growth on hygromycin. All validated mutants were designated an OAL# (Table S1) for identification. The orf of ppe64 containing its native promoter and ribosome-binding site was cloned into pDM101 or pML1335 vectors to generate the episomal (pDM103) or integrative (pDM106) mycobacterial expression vectors, respectively. pOAL301, expression vector for purification of PPE64, was constructed in our previous study (41). The orf of ppsA containing its native ribosome-binding site was cloned into pDM101 to generate the episomal pOAL410 mycobacterial expression vector.
Growth experiments for determining iron utilization
Unless specified, all liquid cultures were grown in sealed square PETG bottles with shaking at 120 rpm, and all incubation was done at 37°C and all liquid and solid growth medium experiments were performed in triplicate. Strains were first grown to the mid-exponential phase in complete 7H9, then washed in sterile PBS containing tyloxapol, and then iron-depleted for five generations in iron-free 7H9 medium containing ADS, glycerol, CAA, and tyloxapol. This iron-depletion protocol was strictly performed before all growth experiments. Iron-depleted cells were passed through a 5.0 μM filter to obtain a single-cell suspension, which was then used to inoculate iron-free liquid 7H9 or plate on iron-free solid 7H10 agar containing specific iron sources as mentioned in the main text. For plating on solid agar plates, single-cell suspension was prepared at an optical density (OD)600 of 0.05, which was serially diluted, and 5 µL of each dilution (specified in the figure) was spotted on agar plates. Bovine serum albumin was added to the medium to a final concentration of 0.5% wt/vol (75 µM) for albumin growth experiments. Unless specified, all Hm medium (liquid and solid) in our study contained 20 µM of the iron chelator DIP. For growth curve experiments (Fig. 1A through E, 5B and 6A), strains were inoculated in 30 mL of medium at an initial OD_600_ of 0.01. For endpoint growth experiments (Fig. 2B and C, 6B), strains were inoculated in a final volume of 200 µL in 96-well plates at an initial OD_600_ of 0.001 in wells containing varying concentrations of Hm. OD of liquid cultures was measured using a BioTek Synergy plate reader. All experiments were performed with a minimum of three biological replicates.
Assessing Hm levels by hemochromogen assay, Hm biosensor, and fluorescence microscopy
Strains were grown to the mid-log phase in complete 7H9, iron-depleted, and then inoculated at an OD_600_ of 1.0 into 30 mL of iron-free 7H9 containing either 10 µM Hm or 10 µM Hm and 75 µM albumin. Strains were harvested after 48 h, and Hm level was determined by hemochromogen assay (45). Briefly, cells were first resuspended in 30 mL of 1× PBS, lysed by sonication, then clarified by low-speed centrifugation at 1,500 × g for 5 min to collect the whole-cell lysate (WCL), and total protein was quantified by BCA assay. To extract Hm, 500 µL of WCL was added to a cuvette into which 0.2 M NaOH, 40% pyridine, and 200 µM potassium ferricyanide were added in 500 µL. Released Hm was then reduced by adding 10 µL of 0.5 M DTT, and absorbance was measured at 557 nm. Hm was quantitatively determined using the extinction coefficient 32.4 mM^−1^ cm^−1^ and then normalized to total protein amount WCL.
The hs1-M7A Hm biosensor expression vector pYUB1874 (46) was transformed in wt, and Δppe64 and transformant were selected on 7H10 Kan agar plates. Iron-depleted strains were inoculated at an OD_600_ of 1.0 in triplicate into 10 mL of albumin-free iron-free 7H9 containing 10 µM Hm, and cells were harvested for temporal analysis. Biosensor green fluorescence (GF) from green fluorescent protein (eGFP) was monitored by excitation at 480 nm and emission at 510 nm, normalized to OD of cultures, and reported relative to day 1. OD and fluorescence were measured using a BioTek Synergy plate reader. GF from Hm biosensor strains was also analyzed by microscopy.
Assessing susceptibility to GaPIX toxicity
Strains were first grown to the mid-log phase in complete 7H9 and then iron-depleted as usual. Iron-depleted cells were passed through a 5.0 μM filter to obtain a single-cell suspension, which was then inoculated in a final volume of 200 µL in 96-well plates at an initial OD_600_ of 0.001 in 7H9 medium containing 1 µM ferric citrate and varying concentrations of GaPIX. OD of cultures was measured using a BioTek Synergy plate reader on day 7. All experiments were performed with a minimum of three biological replicates.
Electron microscopy analysis
For transmission electron microscopy (TEM), cells were fixed in 2% glutaraldehyde in 0.1 M Na Cacodylate for a minimum of 2 h. After three washes (180 mM sucrose in 60 mM Na Cacodylate), cells were fixed in 1% OsO4 (aqueous) for 1 h, followed by three washes (180 mM sucrose in 60 mM Na Cacodylate) and then dehydration in increasing concentrations of ethanol. Cells were washed 3× in propylene oxide as a transitional solvent and then infiltrated with a 1:1 mixture of propylene oxide and EMbed 812 resin. After the removal of propylene oxide, cells were embedded in 100% EMbed812. Thin sections (80–90 nm thick) were stained with uranyl acetate and lead citrate and then viewed with JEOL JEM2100 TEM. For scanning electron microscopy (SEM), cells were fixed in 2% glutaraldehyde in 0.1 M Na Cacodylate for a minimum of 2 h. After three washes (180 mM sucrose in 60 mM Na Cacodylate), cells were placed on poly-L-lysine-coated coverslips. Cells on coverslips were then fixed in 1% OsO4 (aqueous) for 1 h, followed by three washes (180 mM sucrose in 60 mM Na Cacodylate) and then dehydration in increasing concentrations of ethanol. Samples were washed 2× in hexamethyldisilazane for 5 min each and allowed to air dry. Samples were then coated with Au/Pd and imaged in an FEI Quanta 600 FEG SEM.
Ethidium bromide accumulation assay
Strains were first grown to the log phase in 30 mL of complete 7H9 medium and then filtered through a 5.0 μM filter to obtain a single-cell suspension. Cells were then harvested by low-speed centrifugation at 1,500 × g for 10 min and resuspended to a final OD_600_ of 1.0 in uptake buffer (76 mM (NH_4_)2_SO_4, 0.5 M KH_2_PO_4_, 1 mM MgSO_4_, 0.4% glucose, and 0.05% Tween-80). For all strains, 100 μL of cells was added in triplicate in a 96-well plate, and ethidium bromide (EtBr) was then added to a final concentration of 20 µM. Fluorescence was measured by excitation at 530 nm and emission at 590 nm at 1-min interval for 60 min.
Lipid extraction and thin layer chromatography
Lipid extractions were performed following established protocols (47–50). For all lipid extractions, all strains were first grown to an OD_600_ of 1.0 in 50 mL of complete 7H9 medium. For extraction of total apolar lipids, cells were harvested by centrifugation, and the pellet was resuspended by adding 2 mL of methanol-0.3% NaCl (10:1, vol/vol) solution and 1 mL of petroleum ether and then mixed on an end-over-end rotor for 30 min. The sample was then centrifuged at 4,000 × g for 5 min, and the upper layer containing phthiocerol dimycocerosates (PDIMs) was collected in a fresh tube. Another 1 mL of petroleum ether was added to the bottom layer, mixed, centrifuged again, and the upper layer was collected. Upper layer fractions were combined, dried, and then samples were spotted onto thin layer chromatography (TLC) plates and resolved in petroleum ether-diethyl ether (90:10, vol/vol). Strains were also labeled with ^14^C-propionate, and PDIMs were detected by autoradiography for 72 h using a Typhoon Phosphor Screen. For the extraction of surface lipids, cell pellets were resuspended in 5 mL of hexanes and mixed on an end-over-end rotor for 5 min. Samples were centrifuged at 3,000 × g for 5 min, hexane-extracted lipids were collected in a fresh tube. An equal amount of chloroform-methanol (2:1, vol/vol) was added. Samples were dried, resuspended in chloroform-methanol (2:1, vol/vol), and then spotted onto TLC plates. TAG was resolved by TLC in toluene-acetone (80:20, vol/vol). TMM, TDM, and free mycolic acids (FM) were resolved by TLC in chloroform-methanol-water (90:10:1, vol/vol/vol). All TLC plates were dipped in 10% molybdophosphoric acid in ethanol, and lipids were then visualized by charring plates.
Biofilm growth assays
Strains were first grown to the mid-log phase in complete 7H9 and then iron-depleted as usual. Iron-depleted cells were passed through a 5.0 μM filter to obtain a single-cell suspension, which was then inoculated in 24-well plates at a final OD_600_ of 0.01 in iron-free Sauton’s medium containing either 10 µM ferric citrate or 10 µM Hm. Plates were sealed with parafilm, wrapped in aluminum foil, and then incubated static at 37°C. After 5 weeks, media was carefully aspirated using a 25G needle, 1 mL of 1% crystal violet (CV) was added to each well and incubated at 37°C for 10 min. CV was then removed from the wells, biofilm was washed gently 2× with PBS, and 1 mL of 95% ethanol was added to the wells. CV was extracted for 10 min at 37°C, and biofilm mass was then quantified by measuring absorbance at 590 nm. For planktonic growth experiments in Sauton’s medium (Fig. 5C), all protocols were performed exactly as described for 7H9 medium, except tyloxapol was added to a final concentration of 0.02% in Sauton’s medium.
Van10-P vancomycin susceptibility assay
Strains were first grown to the mid-log phase in complete 7H9 supplemented with 0.1 mM propionate and passed through a 5.0 μM filter to obtain a single-cell suspension. Cells were then inoculated in a final volume of 200 µL in 96-well plates at a final OD_600_ of 0.005 in complete 7H9 medium containing 10 µg/mL vancomycin. Cells were incubated for 10 days, and then viability was measured by Alamar Blue assay. The growth percent of strains in vancomycin was determined relative to the growth of strains in the absence of vancomycin.
Purification and refolding of PPE64 and analysis of channel activity in lipid bilayers
The ppe64 expression vector pOAL301 was used to purify recombinant PPE64 from E. coli BL21. PPE64 was purified under denaturing conditions, isolated by nickel chromatography, and refolded in different detergents exactly as we did in our previous study (41). Bilayer experiments were performed using very similar instrumentation and methods as described by Zakharian et al. (51). Synthetic diphytanoyl phosphatidylcholine (DphPC, Avanti Polar Lipids, Birmingham, AL) was used to form planar lipid bilayers. Lipids were solubilized in n-Decane at 20 mg/mL, and a glass capillary tube was used to paint a bilayer in an aperture of 200 µM diameter in a Delrin cup (Warner Instruments, Hamden, CT). The bilayer was painted between an aqueous solution of 1 M KCl, 10 mM HEPES, pH 7.1, and capacitance was registered in the range of 66–100 pF. Approximately ~40 ng of purified detergent refolded PPE64 in 1–2 µL volume was added to the cis compartment, and channel-forming activity was recorded at 50 mV applied potential. Current trace was recorded with a patch clamp amplifier (BC-535 Bilayer Clamp, Warner Instruments). The trans and cis solutions were connected to the headstage point with Ag-AgCl electrodes. Currents were low-pass filtered at 10 kHz and then digitized through an analog-to-digital converter (Digidata 1550B, Molecular Devices, San Jose, CA). Data filtering was done at 100 Hz through an 8-pole Bessel Filter (Lpf-8, Warner Instruments) and digitized at 1 kHz using pClamp11 software (Molecular Devices). Single-channel conductance events were identified automatically using Clampfit 11 from five independent membrane recordings.
Absorption spectroscopy for detecting Hm binding
Fresh solutions of hemin were prepared in Tris buffer as described previously (52). An equimolar amount of Hm was added to 10 μM protein and incubated at room temperature for 5 min. For difference absorption spectroscopy, Hm binding was monitored using a Bio-Tek Synergy HT plate reader by subtracting the free Hm spectra from the protein-incubated Hm spectra.
Cell culture of THP-1 and generation of alveolar macrophage-like cells from human peripheral blood mononuclear cells
THP-1 cells (ATCC# TIB-202) were cultured in base RPMI media (containing 1% L-glutamine and 0.1% BME, 0.005% MEM essential and non-essential amino acids) supplemented with 10% FBS. This medium is referred to as complete RPMI, which was filter-sterilized through a 0.22 µm filter. THP-1 cells were incubated in T-75 flasks at 37°C in 5% CO_2_ and were split according to the manufacturer’s instructions. Prior to use, cells were harvested and quantified using a hemacytometer with trypan blue exclusion dye.
Alveolar macrophage-like cells (AMLs) were differentiated from human peripheral blood mononuclear cells (PBMCs) as previously described (53). Briefly, frozen de-identified human PBMCs (Charles River Laboratories) were thawed and seeded at 6 × 10^6^ cells/well in filter-sterilized RPMI 1640 media with 10% pooled human serum in a six-well ultra-low attachment tissue culture plate. 100 µg/mL Infasurf (ONY Biotech), 10 ng/mL GM-CSF, 5 ng/mL TGF-β, and 5 ng/mL IL-10 (Peprotech) were added at days 0, 2, and 4. On days 2 and 4, 1 mL of fresh media was added to each well after aspirating 1 mL of spent media. On day 6, cells were dissociated with Versene cell dissociation reagent (Gibco) and gentle shaking on an orbital shaker. The cell purity was verified by flow cytometry using HLA-DR and CD11c and evaluated on a NovoCyte 3000 and analyzed using NovoExpress Software (Agilent). Cell concentration was quantified with trypan exclusion dye in a hemacytometer.
Infection assays with THP-1 and AMLs
The day before infection with Mtb strains, THP-1 cells were seeded in 96-well plates at 10^5^ cells/well in 100 µL RPMI containing 50 nM PMA (phorbol 12-myristate 13-acetate), and cells were then differentiated into macrophages overnight. AMLs were similarly seeded into 96-well plates at 10^5^ cells/well in 100 µL RPMI. AML RPMI medium contained Infasurf, GM-CSF, and TGF-β to sustain the AML phenotype. The following day, macrophage monolayers in all wells were washed 2× with sterile PBS, 100 µL of infection medium (RPMI supplemented with 10% non-heat-inactivated normal human serum) was added to all wells, and then Mtb cells in 20 µL sterile PBS were added to all wells at an MOI of 10 and an MOI of 2 for THP-1 and AML infection, respectively. All Mtb strains were first grown to the mid-log phase in complete 7H9, and cells were then passed through a 25G needle to obtain a single-cell suspension for infecting macrophages. Infected monolayers were incubated at 37°C in 5% CO_2_ for 3 h and then washed 2× with sterile PBS to remove extracellular bacteria, and then 100 µL of RPMI containing 10 µg/mL gentamicin was added to all wells. Medium was then removed from wells at indicated timepoints, macrophages were lysed with 100 µL 0.05% SDS, and Mtb was then enumerated by plating on 7H10 agar plates.
Identification of suppressor mutations
We performed whole-genome sequencing of wt, Δppe64, and Δppe64sup at Molecular Biology and Cytometry Research at the OUHSC core facility using NextSeq 2000. We processed the raw reads using fastp v0.23.2 (54) with default settings. We then used snippy v4.6.0 (55) with default settings to identify mutations in these strains, compared to the reference H37Rv genome (GenBank: AL123456.3). To identify probable suppressor mutations, we directly compared the list of single-nucleotide polymorphisms in the Δppe64sup and parental Δppe64 strain.
RESULTS
PPE64 is critical for Hm utilization by the non-albumin Hm uptake pathway
In our previous study (41), we discovered that Mtb H37Rv significantly upregulates the expression of the gene ppe64 in the presence of Hm iron. Subsequently, we characterized PPE64 in binding assays, membrane localization, and channel activity experiments with specific controls to demonstrate that PPE64 binds Hm and most consequentially that PPE64 is localized in the outer mycomembrane and forms water-filled channels (41). These observations suggested that the PPE64 channel protein may play a role in Hm iron utilization in Mtb. To determine whether PPE64 is required for Mtb Hm utilization, we used homologous recombination to construct an unmarked Δppe64 deletion strain (Fig. S1 and Table S1). We first monitored the growth of Δppe64 in standard 7H9 liquid medium, which contains 150 µM ferric citrate (FeCi), supplemented with glycerol and ADS (albumin, dextrose, salt). Under these conditions, wt and Δppe64 exhibit nearly identical growth (Fig. 1A), ensuring that Δppe64 does not have any generalized growth defect. As Mitra et al. (29) have shown that Mtb has two Hm uptake pathways, termed albumin and non-albumin, we next examined whether PPE64 is required for either of these pathways. We monitored the growth of Δppe64 to utilize albumin-Hm by growing strains in iron-free 7H9 supplemented with glycerol and albumin containing either 10 µM FeCi (Fig. 1B) or 10 µM Hm (Fig. 1C). Growth of wt and Δppe64 is nearly identical under these conditions, demonstrating that PPE64 is not required for Hm utilization by the albumin pathway. We next monitored the growth of Δppe64 to utilize non-albumin Hm by growing strains in iron-free 7H9 supplemented with glycerol containing either 10 µM FeCi or 10 µM Hm. In the absence of albumin, Δppe64 exhibits only a marginal growth delay in FeCi (Fig. 1D), but deletion of ppe64 significantly reduces the growth of Mtb in the presence of Hm after day 6 (Fig. 1E). The initial growth for 6 days as observed for Δppe64 in Hm medium has also been observed in other Mtb Hm utilization mutants in our previous studies (29, 30, 41), and this background growth was attributed to either utilization of residual iron from the medium or residual internal iron reserves in the cells of the Mtb strains. Regardless, the growth of Δppe64 is fully restored to wt levels through complementation from the episomal expression vector pDM103 (Table S2), which expresses ppe64 using its native promoter. The Hm growth phenotype of Δppe64 is also recapitulated by plating for CFU on self-made iron-free 7H10 solid agar plates containing either 10 µM FeCi or 10 µM Hm. Since growth on agar plates is significantly slower than in liquid media and because our complement strain OAL154 (Table S1) requires hygromycin, for agar plate assays, we used wt and Δppe64 strains expressing the empty expression vector pDM101 (Table S2) to control for any differences in growth rates. Quantifying CFU shows nearly identical growth of wt and Δppe64 in FeCi, whereas the growth of Δppe64 is reduced 85% compared to wt in the presence of Hm (Fig. 1F; Fig. S2). Altogether, these observations demonstrate that the PPE64 channel protein plays a critical role in Hm utilization by the non-albumin pathway.
PPE64 is critical for Hm iron utilization by the non-albumin Hm uptake pathway. (A ) Growth of wt (black) and Δppe64 (blue) strains in standard liquid 7H9 medium which contains 150 µM ferric citrate (FeCi) and has been supplemented with 1% glycerol, 10% ADS, and 0.02% tyloxapol. (B and C) Growth of strains in iron-free liquid 7H9 medium containing 1% glycerol, 0.02% tyloxapol, and 75 µM albumin, which has been supplemented with either 10 µM FeCi (B) or 10 µM Hm (C).(D and E) Growth of wt, Δppe64, and complement (pink) strains in iron-free liquid 7H9 medium containing 1% glycerol and 0.02% tyloxapol, which has been supplemented with either 10 µM FeCi (D) or 10 µM Hm (E). Inset in E shows triplicate cultures of growth experiment at day 16. (F) Growth of strains on self-made iron-free solid 7H10 agar plates supplemented with 1% glycerol, 10% ADS, and 0.02% tyloxapol containing either 10 µM FeCi or 10 µM Hm. Single-cell suspension of iron-depleted strains was normalized to an OD600 of 0.05, then serially diluted, and 5 µL of each dilution was spotted on agar plates. FeCi and Hm agar plates were imaged on day 21 and 35, respectively, and CFU counts were determined from either 10−3 or 10−4 dilution. The right panel shows a representative image of plates used to determine CFU counts. Uncropped plate images are shown in Fig. S2. All Hm medium contains 20 µM of the iron chelator DIP to prevent utilization of trace iron. All error bars represent the standard error of mean (SEM) values of biological triplicates. In many cases, error bars are smaller than the marker data points. Asterisk denotes Δppe64 is significantly different from wt. Statistical significance was determined by Tukey’s HSD following an F-test (P < 0.05). The source data file is provided.
The PPE64 channel protein is required for Hm uptake in Mtb
Since the PPE64 channel protein is required for growth in Hm, we hypothesized that PPE64 may affect intracellular Hm levels in Mtb. For example, a lack of Hm uptake in Δppe64 would result in the absence of iron nutrients for the cell, or the accumulation of intracellular Hm in Δppe64 could result in Hm toxicity, where either mechanism could lead to the absence of growth in Hm as observed in Δppe64. First, we directly quantified intracellular Hm levels using the pyridine hemochromogen assay (45). Conceptually, harvested cells are first lysed to release Hm, which is reduced to its ferrous iron state and then bound by pyridine. The specific absorbance of the reduced-Hm-pyridine complex at 557 nm can then be monitored to determine Hm levels. Using this assay, we determined that deleting ppe64 significantly lowers intracellular Hm levels in Mtb and that this could be fully restored to wt levels by complementation (Fig. 2A). As an additional control, we also determined Hm levels in strains when grown in the presence of albumin and Hm. Interestingly, albumin significantly stimulates Hm uptake in Mtb as Hm levels in wt were ~2-fold higher when compared to the non-albumin condition (Fig. 2B). Since Δppe64 grows in Hm in the presence of albumin (Fig. 1C), expectedly, there was no significant difference in Hm levels between wt and Δppe64. These results are consistent with the observations that PPE64 is critical for Hm utilization by the non-albumin pathway but has no role in the albumin pathway.
PPE64 influences Hm uptake in Mtb. (A ) Quantification of Hm levels in wt (black), Δppe64 (blue), and complement (pink) strains by hemochromogen assay. Iron-depleted strains were inoculated into iron-free liquid 7H9 medium supplemented with either 10 µM Hm or 10 µM Hm and albumin (Hm + A). Hm levels in strains were quantified after 48 h by the pyridine hemochromogen assay and then normalized to the total protein amount in whole-cell lysates. (B) Growth of strains in iron-free liquid 7H9 medium containing varying amounts of the toxic Hm analog gallium-protoporphyrin IX (GaPIX). To allow for the background level of growth, 1.0 µM FeCi was included as an iron source. Growth was determined by measuring endpoint OD 750 on day 7. (C) Growth of strains in iron-free liquid 7H9 medium containing increasing levels of Hm. Strains were grown in 96-well plates, and growth was determined by measuring endpoint OD 750 on day 35. The right panel shows images of 96-well plates. (D) Determination of Hm levels using a cytoplasmic Hm biosensor. Green fluorescence (GF) from cytoplasmic Hm biosensor in strains grown in iron-free liquid 7H9 supplemented with 10 µM Hm. GF from Hm biosensor was monitored at 24-h intervals, normalized to OD 750, and is reported relative to day 0. (E) Hm biosensor fluorescence in strains examined by microscopy. GF was monitored at the same exposure levels for both strains. Raw image files are provided. All error bars represent the SEM of biological triplicates. For A, plots with different lowercase letters indicate statistically significant differences from each other. Asterisk denotes Δppe64 is significantly different from wt. Statistical significance was determined by Tukey’s HSD following an F-test (P < 0.05). Source data file is provided.
The reduced intracellular Hm levels in Δppe64 suggested that PPE64 may influence Hm uptake in Mtb. To test this hypothesis, we examined the susceptibility of Δppe64 to the toxic Hm analog GaPIX. We have previously (30) shown that GaPIX uptake is mediated by Hm uptake components because of their structural similarity. Thus, we reasoned that if Δppe64 has reduced Hm uptake, then it would be less susceptible to GaPIX toxicity due to reduced GaPIX uptake. Conversely, if intracellular Hm is accumulating in Δppe64, then it would be more susceptible to GaPIX toxicity due to accumulating GaPIX. We observed that the deletion of ppe64 increases Mtb resistance to GaPIX, and susceptibility is fully restored upon complementation (Fig. 2B), which suggests that ppe64 deletion reduces GaPIX uptake. We also determined the growth pattern of Δppe64 in the presence of increasing levels of Hm. Since excess Hm can be toxic to cells, we reasoned that if Δppe64 accumulates Hm, then Δppe64 growth would not recover in the presence of higher levels of Hm due to increasing Hm accumulation and toxicity. Increasing the Hm concentration to 20 µM and 40 µM has no effect and does not recover Δppe64 growth (Fig. 2C). However, Δppe64 growth recovers nearly 50% at 80 µM Hm and to wt levels at 160 µM Hm (Fig. 2C), suggesting that Δppe64 is not experiencing Hm toxicity.
Finally, we monitored intracellular Hm levels using the HS1-M7A cytoplasmic Hm biosensor, which we previously used in our study to monitor Hm uptake (41). This biosensor has the Hm binding domain of cytochrome b562 conjugated to a green fluorescent protein (56). During regular Hm uptake, Hm is transported into the cell, and Hm binding by b562 quenches green fluorescence (GF), whereas during defective Hm uptake, Hm transport into the cell is reduced or absent, and GF is not quenched. The Hm biosensor expression vector pYUB1874 was transformed into wt and Δppe64 strains, and GF was monitored temporally at 24-h intervals. We observed that the GF steadily and significantly increases in Δppe64 over time, indicating that Hm levels are lower in Δppe64 compared to wt (Fig 2D). GF in Δppe64 peaked at ~3-fold higher levels compared to that in wt by day 5, which was visually validated through microscopy analysis (Fig. 2E). Collectively, all of our observations present a convincing case that PPE64 plays a critical role in Hm uptake by the non-albumin Hm utilization pathway in Mtb.
Absence of PPE64 triggers pleiotropic effects on Mtb physiology
During our microscopic analysis of the Hm biosensor strains, we observed that the Δppe64 cells appeared elongated compared to the wt cells. To further validate these observations, we cultured Mtb strains in standard 7H9 liquid medium supplemented with glycerol and ADS and analyzed them by both TEM and SEM (Fig. 3A). WT displayed a cell length range of 1.2–2.5 µm with an average length of 1.9 µm (Fig. 3B). However, deleting ppe64 increases Mtb cell length range to 1.2–4.2 µm with an average length of 2.7 µm with an overall rougher cellular morphology. Interestingly, overproduction of PPE64 in the complement strain significantly reduces the average cell length to 1.5 µm and results in smoother cellular morphology. Since PPE64 is a mycomembrane channel protein that also affects Mtb cell shape, we examined whether Δppe64 has altered cell permeability, as this can affect the trafficking of hydrophobic molecules such as Hm. Monitoring membrane permeability by measuring EtBr accumulation (57) showed that deleting ppe64 reduces the rate of EtBr accumulation, suggesting that Δppe64 has reduced cell membrane permeability (Fig. 4A).
PPE64 influences Mtb cellular morphology. (A ) Analysis of wt, Δppe64, and complement strains by TEM and SEM. Strains were grown in standard liquid 7H9 supplemented with 1% glycerol, 10% ADS, and 0.02% tyloxapol. Full uncropped images are provided in Fig. S3. (B) Quantification of cell size of wt, Δppe64, and complement strains from electron microscopy. Three different SEM images for each strain were analyzed in ImageJ, and at least 30 cells for each strain were analyzed to determine cell size. Plots with different lowercase letters indicate statistically significant differences from each other as determined by Tukey’s HSD following an F-test (P < 0.002). The source data file is provided.
PPE64 influences Mtb cell membrane permeability. (A) Cell permeability in wt (black), Δppe64 (blue), and complement (pink) strains was monitored by assessing EtBr accumulation. The source data file is provided (B–D). Analysis of lipids in Mtb strains by TLC. (B) Total apolar lipid extracts were resolved by TLC in petroleum ether-diethyl ether (90:10 vol/vol). (C) Surface lipids were resolved by TLC in toluene-acetone (80:20 vol/vol) to visualize TAG. TLC experiments were performed with two biological replicates. (D) Surface lipids were resolved by TLC in chloroform-methanol-water (90:10:1 vol/vol/vol) to visualize TMM, TDM, and FM. “a” denotes an unknown lipid. Results of biological replicates are shown in Fig. S4. (PDIM: pthiocerol dimycocerosates, FM: free mycolic acids, TDM: trehalose dimycolates, TMM: trehalose monomycolates, TAG: triacylglycerol).
As it is well-established that mycomembrane lipids play a major role in membrane permeability (48, 58), we next examined the cell surface lipid profile of Δppe64. Strains were cultured similarly, as done for permeability experiments, and apolar and surface lipids were extracted and examined by TLC. Total apolar lipid extracts were resolved in a petroleum ether-diethyl ether solvent system to visualize PDIMs, a major lipid factor that influences Mtb membrane permeability. In Mtb, during PDIM biosynthesis, two mycocerosates are esterified onto phthiodolone to produce the first PDIM product called PDIM B, which is further reduced and methylated to produce PDIM A (59). Our TLC results from two independent biological experiments show a trend where ppe64 deletion slightly reduces PDIM A levels (Fig. 4B). Furthermore, we resolved hexane-extracted surface lipids in a toluene-acetone and chloroform-methanol-water solvent systems to visualize triacylglycerol (TAG), FM, and trehalose mono/dimycolates (TMMs, TDMs). While deletion of ppe64 does not affect TAG (Fig. 4C; Fig. S4B) or FM/TMM/TDM levels (Fig. 4D; Fig. S4C), in the chloroform-methanol solvent system, we observed the loss of an unknown lipid (Fig. 4D; Fig. S4C). Production of this surface-extractable lipid (denoted as “a”) was again restored to wt levels upon complementation. Collectively, our data suggest an important role of the PPE64 channel protein in somehow maintaining cell shape and membrane permeability in conjunction with influencing levels of certain mycomembrane cell surface lipids.
PPE64 moonlights in Hm iron utilization and biofilm formation
There is compelling evidence that variations in iron (60) and cell surface lipids (10, 61) levels can have significant effects on mycobacterial biofilm formation. Since we observed that deleting ppe64 affects Hm iron utilization and levels of some mycomembrane lipids, this prompted us to assess whether PPE64 plays any role in Mtb biofilm formation. Biofilms were grown for 5 weeks in typical (62) albumin-free detergent-free Sauton’s medium, and biofilm mass was determined by CV staining, but with one specific modification: strains were grown in iron-free Sauton’s containing either 10 µM FeCi or 10 µM Hm to assess the effect of specific iron sources. From the biofilm mass measurements and direct visual examination, it was apparent that wt Mtb forms more robust biofilms when Hm is available as an iron source compared to the non-Hm iron ferric citrate (Fig. 5). Based on our observation that Δppe64 cannot utilize Hm iron (Fig. 1E), we expected Δppe64 would not form biofilms in the presence of Hm as the sole iron source. Surprisingly, Δppe64 fails to form the typical pellicular biofilm either in the presence of Hm iron or ferric citrate iron (Fig. 5A), which was fully restored to wt levels upon complementation. This biofilm defect in Δppe64 was specific to the pellicle formation at the air-liquid interface, as visual examination clearly showed growth at the bottom of the wells (Fig. S4D). To ensure that the absence of biofilm in Δppe64 is not a medium-specific effect, we examined the phenotype of all strains grown planktonically in standard Sauton’s medium, which contains ~150 µM ferric citrate, supplemented with tyloxapol. Deleting ppe64 causes a slight delay in growth in Sauton’s medium (Fig. 5B) similar to what is observed in albumin-free 7H9 medium containing ferric citrate (Fig. 1D). In these growth experiments, statistically significant differences between wt and Δppe64 were observed on days 12, 18, and 22 (Fig. 5B). However, when CFU was enumerated from these timepoints, we did not observe any statistically significant differences (Fig. 5C). These observations demonstrate that while Δppe64 exhibits a slight growth delay under planktonic growth in Sauton’s, it exhibits a significant impairment in forming biofilms when grown statically in Sauton’s medium.
PPE64 influences Mtb growth within biofilm independent of the iron source. (A) Quantification of biofilm mass in wt (black), Δppe64 (blue), complement (pink), and Δppe64 suppressor (cyan) strains grown in detergent-free iron-free Sauton’s medium supplemented with either 10 µM FeCi or 10 µM Hm. Biofilms were grown for 5 weeks in 24-well plates and quantified by CV staining. The right panel shows an image of the plate used for biofilm quantification. Plots with different lowercase letters indicate statistically significant differences from each other. (B) Growth of wt (black), Δppe64 (blue), complement (pink), and Δppe64 suppressor (cyan) strains in standard Sauton’s liquid medium (which contains 150 µM FeCi) supplemented with 0.02% tyloxapol. (C) Growth of strains in liquid Sauton’s medium at the indicated time points was monitored by determining bacterial CFU counts by plating on 7H10 agar. All error bars represent the SEM of biological triplicates. Asterisks denote that Δppe64 is significantly different from wt at those timepoints. Timepoints with black lowercase letters indicate that there were significant differences between Δppe64 and Δppe64 suppressor strains at those timepoints. Statistical significance was determined by Tukey’s HSD following an F-test (P < 0.05). The source data file is provided.
In our experiments, while characterizing the growth of Δppe64 in Hm, we made a serendipitous observation, where after ~70 days, Δppe64 abruptly started growing in the non-albumin 7H9 medium containing Hm. This suggested that Δppe64 may have accumulated a suppressor mutation allowing it to start utilizing Hm again in the non-albumin condition. We isolated a single suppressor strain (denoted Δppe64sup) and in growth experiments, verified that this strain is fully capable of utilizing Hm under non-albumin conditions (Fig. 6A) and generally outperforms wt when grown in different concentrations of Hm (Fig. 6B; Fig. S5A). In hemochromogen assays, we validated that Δppe64sup has significantly high levels of intracellular Hm compared to both wt and Δppe64 (Fig. 6C), which is consistent with its recovered Hm growth phenotype. Identification of this Δppe64sup Hm suppressor mutant presented a unique opportunity to further examine any connections between the Hm iron utilization and biofilm formation roles of PPE64. We similarly characterized Δppe64sup in biofilm assays and observed that the recovered Hm-utilizing phenotype in the suppressor does not recover the ability of Δppe64 to form biofilms (Fig. 5A). Moreover, Δppe64sup also outperforms all other strains when grown planktonically in Sauton’s medium (Fig. 5B and C). These observations convincingly demonstrate that the functions of PPE64 in Hm iron utilization and biofilm formation are discrete and unlinked.
A suppressor mutation restores the ability of the ppe64 mutant to utilize Hm. (A) Growth of wt (black) and Δppe64-suppressor mutant (cyan) strains in iron-free liquid 7H9 medium supplemented with 10 µM Hm. (B) Growth of wt (black), Δppe64 (blue), and Δppe64-suppressor (cyan) strains in iron-free liquid 7H9 medium containing varying concentrations of Hm. Strains were grown in 96-well plates, and growth was determined by measuring endpoint OD750 on day 20. Full uncropped plate images are shown in Fig. S5A. (C) Quantification of Hm levels in strains by hemochromogen assay. (D) Cell permeability in strains was monitored by assessing EtBr accumulation. (E) Survival of strains in standard liquid 7H9 medium supplemented with 1% glycerol, 10% ADS, and 0.02% tyloxapol in the presence of 10 µg/mL vancomycin determined by the microplate Alamar Blue assay. The growth percent of strains in vancomycin was determined relative to the growth of strains in the absence of vancomycin. (F) Analysis of PDIM by TLC in strains labeled with 14C-propionate demonstrating the presence or absence of PDIMs. (G) Growth of strains in iron-free liquid 7H9 medium containing 1% glycerol, 0.02% tyloxapol, and 10 µM Hm. (H) Growth of strains on self-made iron-free solid 7H10 agar plates supplemented with 1% glycerol, 10% ADS, 0.02%, and 10 µM Hm (n = 5). The right panel shows a representative image of plates used to determine CFU counts. Uncropped plate images are shown in Fig. S5D. All error bars represent the SEM of a minimum of biological triplicates. For C, E, and H, plots with different lowercase letters indicate significant differences (P < 0.04). Statistical significance was determined by Tukey’s HSD following an F-test (P < 0.05). The source data file is provided.
Reducing PDIM levels increases Hm uptake
To establish a possible mechanism for how Δppe64 could utilize Hm again, we performed further phenotypic characterization of the suppressor strain. As Δppe64sup has increased intracellular Hm, we first examined its cell membrane permeability by monitoring EtBr accumulation. The EtBr accumulation rate in Δppe64sup was significantly higher compared to wt, Δppe64, and complement strains, suggesting that Δppe64sup has increased membrane permeability (Fig. 6D). As mycomembrane PDIM levels can affect EtBr accumulation rates, we determined PDIM levels in the suppressor strain using two orthogonal methods. First, we performed a Van10-P vancomycin susceptibility assay, which was recently (50) established as a straightforward and highly effective method to assess PDIM levels. In this assay, increased susceptibility to vancomycin strongly correlates with reduced PDIM levels. The Van10-P assay showed that Δppe64sup is far more susceptible to vancomycin compared to all other strains, suggesting that the suppressor has reduced PDIM levels (Fig. 6E). Second, we confirmed the Van10-P observations by directly assessing PDIM levels, which demonstrated that PDIM levels are indeed reduced in the suppressor strain (Fig. 6F). These observations suggested that the suppressor strain may have accumulated mutations that result in reduced PDIMs. As such, using whole-genome sequencing, we identified two non-synonymous (rv1611 and rv2374c), one synonymous (rv3152c), and one frameshift mutation (rv2931) (Fig. S5B and C) in the suppressor mutant strain. Of these four mutations, the most consequential mutation is the frameshift mutation in rv2931, which encodes PpsA, a multifunctional protein that plays a key role in initiating PDIM biosynthesis (63, 64). In Δppe64sup, a single-nucleotide insertion in ppsA results in an opal mutation (premature TGA stop codon, Fig. S5C), suggesting that the production of a truncated PpsA may lead to reduced PDIM levels as observed in Δppe64sup. We hypothesized that reduced PDIM levels in Δppe64sup may allow cells to again traffic Hm, alleviating the Hm utilization defect of Δppe64. If this was the case, then reproducing PDIMs in Δppe64sup should again prevent the cells from utilizing Hm. To test this hypothesis, we overexpressed wt ppsA in Δppe64sup using the episomal vector pOAL410. First, we confirmed that PDIM is reproduced in Δppe64sup by assessing PDIM levels (Fig. S5D) and measuring susceptibility of strains to vancomycin in Van10-P assays (Fig. S5E). Next, we determined the ability of strains to utilize Hm. Expectedly, reproduction of PpsA in the suppressor (Sup ppsA+) prevents Δppe64sup from growing in the presence of Hm (Fig. 6G and H ; Fig. S5F), confirming our hypothesis. Collectively, our observations suggest that reduced PDIM levels in Δppe64sup increase membrane permeability, which allows Δppe64 to again traffic and utilize Hm.
PPE64 has two oligomeric states with different properties
In our previous study (41), we established a detailed process of purifying and refolding the PPE64 mycomembrane protein in the presence of the detergent OPOE (n-Octyl-oligo-oxyethylene). Subsequently, in electrophysiology experiments with planar lipid bilayers with specific controls, we demonstrated that PPE64 forms water-filled channels (41). Our observations showing that PPE64 plays separable roles in Hm iron utilization and biofilm formation suggested that PPE64 may have different protein folded states or domains functioning in these distinct processes, prompting us to further examine the properties of PPE64. We purified recombinant PPE64 and then tested refolding of PPE64 in different detergents such as OPOE (control), DDM (n-dodecyl-β-D-maltoside), and OG (octyl glucoside). While we did not achieve any refolding in OG, PPE64 could be refolded in DDM, similar to OPOE. Analysis of refolded PPE64 by size exclusion chromatography (SEC) showed that PPE64 is refolded into a single oligomeric state in OPOE and in two different oligomeric states in DDM, where all SEC fractions showed the presence of monomeric PPE64 (~62 kDa) as visualized by denaturing PAGE (Fig. 7A; Fig. S6). Analysis of SEC fractions by non-denaturing PAGE confirmed that OPOE refolds PPE64 into a single oligomeric state (fraction F7) of ~480–720 kDa, and DDM refolds PPE64 into two oligomeric states of ~480–720 kDa (F8) and ~146–242 kDa (F10), where the F10 oligomer is the predominant species in DDM (Fig. 7B; Fig. S6). We next examined the channel-forming properties of all PPE64 oligomers in electrophysiology experiments as we have done before (41). We formed a lipid membrane in the aperture of a Delrin cup using DphPC lipids, a specific PPE64 oligomer was added to the cis compartment, and the current trace was temporally monitored. F7 of PPE64_OPOE_ rapidly was inserted into the lipid bilayer and formed channels with an average conductance of ~14 nS (Fig. 7C), which is in line with our previous observations (41). F8 of PPE64_DDM_ inserted into the lipid bilayer at a slower rate and formed channels with an average conductance of ~20 nS (Fig. 7D). In contrast, F10 of PPE64_DDM_ did not form any pores (Fig. S6C). We also determined Hm-binding capability of these PPE64 oligomers through absorption spectroscopy by monitoring the presence of the characteristic Soret peak at ~410 nm, indicative of protein-Hm binding. F7 of PPE64_OPOE_ exhibits a strong Soret peak indicative of Hm binding (Fig. 7E), recapitulating our previous observations (41). F8 of PPE64_DDM_ similarly exhibits a strong Soret peak; however, F10 of PPE64_DDM_ has significantly reduced absorbance at 410 nm indicative of very weak interactions with Hm (Fig. 7E). Collectively, these observations demonstrate that the lipidomic environment can direct the formation of at least two oligomeric states in PPE64 and that the higher-order oligomeric state is responsible for channel activity and interacting with Hm.
The higher-order oligomeric state of PPE64 forms water-filled membrane channels. (A ) Analysis of PPE64 refolded in OPOE (blue) or DDM (pink) by SEC using Sepax SRT-10C SEC 300 column. Colored numbers within the chromatogram show the fraction number. Bio-Rad standard (black dotted line) peaks: a— 670 kDa, b—158 kDa, c—44 kDa, and e—1.3 kDa. The right panel in A shows analysis of SEC fractions by SDS-PAGE. PPE64 monomer is ~64 kDa. (B) Analysis of SEC fractions by NATIVE-PAGE showing two oligomeric states of PPE64. F7 (OPOE) and F8 (DDM): 480-720 kDa; F10 (DDM): 146–242 kDa. (C and D) Channel-forming activity of PPE64 protein fractions in planar lipid bilayers. Proteins were added to DphPC membranes with 50 mV applied potential, and then the current trace was recorded for PPE64-F7OPOE (C), PPE64-F8DDM (D), and PPE64-F10DDM (Fig. S6C). Each stepwise increase (orange arrows) in the current trace represents a protein-mediated channel formation in the lipid bilayer. Y-axis scales are the same in (C–E). Detection of Hm binding by PPE64 SEC fractions through difference absorption spectroscopy. Free heme spectra were subtracted from heme-incubated protein spectra at protein concentrations of 10 µM. The source data file is provided. For (C and D), source data acquisition files (axon binary files) are too large for upload and require pClamp software for viewing and will be provided upon request. Full uncropped images of all protein gels are shown in Fig. S6.
PPE64 is important for Mtb growth in human macrophages
Our observations show that PPE64 affects multiple aspects of Mtb physiology: Hm iron utilization, biofilm formation, cell shape, and membrane permeability. As such, we wanted to determine whether PPE64 contributes to virulence by examining the growth of Mtb within macrophages. For macrophage infection experiments, we constructed a separate Δppe64 complement strain by using the integrative expression vector pDM106 (Table S2), which expresses ppe64 using its native promoter. In infection experiments with THP-1 cells differentiated into macrophages, deletion of ppe64 significantly reduced Mtb survival at 3 and 5 days post-infection, with a trend showing slow recovery in Δppe64 (Fig. 8A). We also examined the growth of Mtb in AMLs generated from human PBMCs. A recent study by the Schlesinger (53) group demonstrated that AMLs derived from human PBMCs serve as an excellent simplified in vivo proxy for studying Mtb infections. We generated high-purity AMLs from human PBMCs and validated purity using flow cytometry by CD11c and HLA-DR staining. We routinely achieved 95%–99% AML purity, as shown in the representative flow cytometry plot (Fig. S6D). A noticeable difference was that AMLs are more permissive to Mtb growth relative to differentiated THP-1 cells. Following 24 h post-infection, while wt Mtb exhibits an initial reduction in growth in THP-1, it proliferates within AMLs (Fig. 8B). This pattern was apparent for all Mtb strains in both infection conditions. Deleting ppe64 also significantly reduced growth of Mtb in AMLs with a similar trend of recovery (Fig. 8B). The ex vivo growth defect of Δppe64 recovered to near wt levels upon complementation. Collectively, these observations demonstrate that under these experimental conditions, PPE64 plays an important role in Mtb survival within human macrophages.
PPE64 influences Mtb growth within macrophages. (A) THP-1 monocytes were differentiated into macrophages with PMA and then infected with Mtb strains at an MOI of 10. Macrophages were lysed at the indicated time points, and Mtb viability was determined by enumerating CFU on agar plates. (B) AMLs were infected with Mtb strains at an MOI of 2. Infected AMLs were lysed at the indicated time points, and Mtb viability was determined by enumerating CFU on agar plates. All error bars represent the SEM of biological triplicates. Asterisk denotes that Δppe64 is significantly different from wt at those timepoints. Statistical significance was determined by Tukey’s HSD following an F-test (P < 0.05). Source data file is provided.
DISCUSSION
Our study started with the goal of characterizing the role of the PPE64 mycomembrane channel protein in Hm iron utilization. We know that Mtb has at least two means of acquiring Hm iron, termed the non-albumin- and albumin-dependent pathways (29). Under the experimental conditions examined in our study, we discovered that PPE64 is a major requirement for Mtb Hm utilization by the non-albumin pathway at Hm concentrations of 10, 20, and 40 µM (Fig. 1 and 2C) and that it is important for Hm uptake (Fig. 2). The requirement for PPE64 also appears to be conditional, because the ppe64 mutant grows when Hm level is increased to 80 and 160 µM (Fig. 2C). Since Hm is highly reactive, free Hm is extremely rare in the human body and is sequestered within hemoproteins. Thus, it is debatable whether Mtb would ever encounter free Hm levels of 80–160 µM under physiologically relevant conditions in the host. But it is conceivable that Mtb may perhaps use a low-affinity Hm transporter(s) at high Hm levels (80–160 µM), whereas PPE64 functions in Hm transport at lower levels of Hm. Alternatively, 80–160 µM Hm may be high enough that Hm overcomes the PDIM barrier in the mycomembrane, bypassing the need for PPE64, allowing Δppe64 to grow. Regardless, our data clearly establish an important role of PPE64 in trafficking Hm into the Mtb cell by the non-albumin pathway.
Through our microscopy analysis, we observed that PPE64 also influences Mtb cellular morphology, where in its absence, cells are more elongated, and PPE64 overproduction leads to smaller cell size (Fig. 3). To the best of our knowledge, this is the first evidence of a PPE protein affecting cell size. It is known that during cell division, mycobacterial cells elongate asymmetrically at the poles, producing daughter cells of different size, growth rate, and cell wall composition (65–68). There is ample evidence demonstrating that the spatiotemporal organization of OMPs and interactions with peptidoglycan play important roles in cell division in diderm bacteria (69–73). Moreover, the outer membrane channel protein OmpA (74) in gram-negative bacteria plays a critical role in interacting with other OMPs and LPS to order and stabilize the outer membrane (70, 75). These observations raise important questions about whether the Mtb PPE64 channel protein could have functions in influencing mycomembrane stability or the mycobacterial cell division apparatus and opens exciting new avenues of research for future studies. We also observed that the absence of PPE64 renders Mtb cells less permeable to EtBr (Fig. 4A), which can sometimes be a consequence of altered lipid levels in the mycomembrane. In our lipidomic analyses, while we observed a trend of slight reduction of PDIM A levels in the ppe64 mutant (Fig. 4B), it must be noted that these differences could be a result of variations in sample loading as observed between the two experimental replicates (Fig. S4A). The trend of reduced PDIM levels is consistent with slightly increased susceptibility to vancomycin as observed in Δppe64 (Fig. 6E). This is in line with recent findings showing that the loss of PDIM increases susceptibility to vancomycin (50). However, the trend of slightly reduced PDIM A in Δppe64 is not due to the random loss of PDIM production that can happen in Mtb cultures in vitro (76, 77), as complementation reproduces PDIM A to wt levels. Furthermore, loss of PDIM increases cell permeability (58) and EtBr accumulation (50), which is not the case in Δppe64, suggesting that the marginal change in PDIM A level is not the cause of reduced EtBr accumulation in Δppe64. In a recent study, Rodrigues et al. demonstrated that loss of the mycomembrane porin MspA reduces EtBr accumulation in M. smegmatis (78). Since PPE64 is a mycomembrane channel protein in Mtb (41) (Fig. 7C and D), it is conceivable that PPE64 could traffic EtBr across the mycomembrane and hence its absence reduces EtBr accumulation in Δppe64. Altogether, our data further establish a link between mycobacterial PPE proteins, PDIM levels, and membrane permeability, which has been shown in previous studies (34, 79). Nonetheless, we made a serendipitous discovery that PDIM levels can influence Hm uptake because the Δppe64 suppressor mutant, which can utilize Hm, produces far less PDIMs compared to wt and Δppe64 (Fig. 6F). Reproducing PDIM levels (Fig. S5D and E) in the Δppe64 suppressor mutant recapitulated the Hm utilization defect (Fig. 6G and H), demonstrating that PDIM levels in the mycomembrane can influence Hm uptake and utilization. It should be noted that we did not undertake a second attempt to isolate a suppressor mutant, and thus we do not know whether reducing PDIMs is a general response to alleviate the loss of Hm utilization phenotype in Δppe64. In our TLC analysis, we also observed that an unknown surface lipid (Fig. 4D, denoted “a”) was absent in Δppe64, and we do not know whether this lipid could influence membrane permeability. A limitation of our lipidomic studies is that analysis was conducted only on strains grown in the typical standard 7H9 growth medium containing albumin. Since PPE64 is essential for Hm utilization in the absence of albumin, a key future study will be to comprehensively characterize the mycomembrane lipid composition of Mtb strains in medium with and without albumin and in different iron sources.
The most consequential finding of our study is that PPE64 has discrete moonlighting functions in Hm iron acquisition and biofilm formation in Mtb. Under planktonic growth conditions, PPE64 is essential for Hm iron utilization by the non-albumin pathway (Fig. 1E). Whereas, under biofilm (static) growth conditions, PPE64 is important for the typical Mtb pellicular biofilm formation (Fig. 5A). The role of PPE64 in biofilm formation is independent of the iron source as the Δppe64 suppressor mutant, which can utilize Hm, remains defective in biofilm formation. This bifunctional nature of the PPE64 channel protein is reminiscent of bifunctional outer membrane channel proteins of Shigella and E. coli that function in discrete processes such as motility, adhesion, and biofilm formation depending on the host niche. For example, the dual-functioning outer membrane channel protein IcsA is crucial for actin-based motility (80) and biofilm formation (81) depending on the stage of the S. flexneri infection cycle. In E. coli, the outer membrane channel proteins AIDA (82) and Ag43 (83) have diverse functions in cell aggregation, cell-to-cell contact, and biofilm formation. Another prime example is the dual functioning outer membrane channel protein OmpM of gram-negative Firmicutes, which is crucial for nutrient uptake and tethering the outer membrane to the cell wall (84). Thus, there is ample precedence to suggest that the distinct functions of PPE64 may also be host niche-specific. For example, macrophages play a key role in Hm recycling (85), and a significant part of the Mtb life cycle is within macrophages. As such, Mtb could be using PPE64 to acquire host Hm iron during intracellular growth. While our observations from infected macrophages (Fig. 8) seem to support this hypothesis, we cannot irrefutably state that the impairment is solely due to loss of Hm acquisition or from other pleiotropic effects in Δppe64. Of importance, we observed that AMLs are more permissive to Mtb growth than THP-1 cells, in line with previous findings (53, 86–89). Since THP-1 monocytes serve as precursors for many subsets of macrophages, this could explain the constrained growth of Mtb in THP-1 macrophages. A limitation of these macrophage experiments is that we can only assess how PPE64 affects intracellular Mtb growth. Ample evidence demonstrates that extracellular Mtb (i) form biofilms in mice, non-human primates, and human lungs, leading to drug tolerance (8), (ii) form biofilms on airway epithelial cells (6), and (iii) clinical Mtb isolates upregulate ppe family genes during biofilm growth (90). Thus, PPE64 may also be an important factor for Mtb biofilm-dependent growth in the host during its extracellular life cycle.
Dissecting the bifunctional roles of PPE64 and their specific contributions to Mtb survival within the host is not a trivial matter, as this requires carefully controlled experiments, use of appropriate infection models, and most importantly, structural knowledge of PPE64. Our biochemical characterization clearly shows that PPE64 (~62 kDa monomer) has at least two oligomeric states (Fig. 7). The higher-order oligomer (~480–720 kDa, predicted nonamer) forms channels in membranes and exhibits strong Hm binding, and thus we hypothesize that this oligomeric state functions in Hm utilization (Fig. 1E) and uptake (Fig. 2). The lower-order oligomer (~146–242 kDa, predicted trimer) does not form channels or bind Hm and may be the oligomeric state that functions in biofilm-dependent growth. We fully recognize that these hypotheses must be validated with detailed structural examination of PPE64 and that the findings of our study only begin to address the many unanswered questions surrounding how mycobacterial PPE proteins function in Mtb physiology. Most importantly, our previous (41) and current studies present conclusive evidence that PPE64 is a novel mycomembrane channel protein that plays major roles in mycobacterial physiology and contributes to Mtb growth ex vivo. The localization of PPE64 in the cell surface, its contribution to Mtb virulence, and its presence only in pathogenic mycobacteria make it an appealing candidate for developing highly targeted chemotherapy. In conclusion, we believe our study presents exciting new findings that will open new avenues of research for the field.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1WHO. 2024. Global tuberculosis report 2024
- 2Murdoch CC, Skaar EP. 2022. Nutritional immunity: the battle for nutrient metals at the host-pathogen interface. Nat Rev Microbiol 20:657–670. doi:10.1038/s 41579-022-00745-635641670 PMC 9153222 · doi ↗ · pubmed ↗
- 3Kurthkoti K, Amin H, Marakalala MJ, Ghanny S, Subbian S, Sakatos A, Livny J, Fortune SM, Berney M, Rodriguez GM. 2017. The capacity of Mycobacterium tuberculosis to survive iron starvation might enable it to persist in iron-deprived microenvironments of human granulomas. m Bio 8:e 01092-17. doi:10.1128/m Bio.01092-1728811344 PMC 5559634 · doi ↗ · pubmed ↗
- 4Waldron KJ, Rutherford JC, Ford D, Robinson NJ. 2009. Metalloproteins and metal sensing. Nature 460:823–830. doi:10.1038/nature 0830019675642 · doi ↗ · pubmed ↗
- 5Sarathy JP, Dartois V. 2020. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin Microbiol Rev 33:e 00159-19. doi:10.1128/CMR.00159-1932238365 PMC 7117546 · doi ↗ · pubmed ↗
- 6Barclay AM, Ninaber DK, Limpens RWAL, Walburg KV, Bárcena M, Hiemstra PS, Ottenhoff THM, van der Does AM, Joosten SA. 2024. Mycobacteria develop biofilms on airway epithelial cells and promote mucosal barrier disruption. i Science 27:111063. doi:10.1016/j.isci.2024.11106339502292 PMC 11536035 · doi ↗ · pubmed ↗
- 7Mishra R, Hannebelle M, Patil VP, Dubois A, Garcia-Mouton C, Kirsch GM, Jan M, Sharma K, Guex N, Sordet-Dessimoz J, Perez-Gil J, Prakash M, Knott GW, Dhar N, Mc Kinney JD, Thacker VV. 2023. Mechanopathology of biofilm-like Mycobacterium tuberculosis cords. Cell 186:5135–5150. doi:10.1016/j.cell.2023.09.01637865090 PMC 10642369 · doi ↗ · pubmed ↗
- 8Chakraborty P, Bajeli S, Kaushal D, Radotra BD, Kumar A. 2021. Biofilm formation in the lung contributes to virulence and drug tolerance of Mycobacterium tuberculosis. Nat Commun 12:1606. doi:10.1038/s 41467-021-21748-633707445 PMC 7952908 · doi ↗ · pubmed ↗