Castleman disease with systemic manifestation: a case report
Ahlam Hmimsa, Nabil Touihem, Hicham Attifi, Mounir Hmidi

TL;DR
This paper reports a case of multicentric Castleman disease in a 65-year-old patient with systemic symptoms and multiple lymph node enlargements.
Contribution
The novelty lies in highlighting Castleman disease as a rare cause of unexplained polyadenopathy with atypical systemic manifestations.
Findings
The patient presented with polyadenopathy and Raynaud's syndrome, leading to a diagnosis of multicentric Castleman disease.
Imaging and histopathological analysis confirmed the diagnosis, emphasizing the need for this condition in differential diagnosis.
The case underscores the importance of considering Castleman disease in patients with systemic symptoms and lymphadenopathy.
Abstract
La maladie de Castleman est une pathologie lymphoproliférative rare. Elle se manifeste par une lymphadénopathie et peut être associée à des manifestations systémiques parfois sévères. Elle peut se présenter sous forme unicentrique, limitée à un seul ganglion ou à une seule région ganglionnaire, et est généralement de bon pronostic. Son traitement repose essentiellement sur l'exérèse chirurgicale, ou multicentrique plus sévère, caractérisée par une atteinte ganglionnaire diffuse et une hyperproduction de cytokines, notamment l'Interleukine-6 (IL-6), expliquant les manifestations systémiques. Le traitement fait appel aux anticorps monoclonaux ciblant l'IL-6, aux immunosuppresseurs, à la chimiothérapie ou à la corticothérapie en fonction de l'étiologie. Elle peut être associée à l'infection par le virus de l'herpès humain type 8 (HHV-8), notamment chez les patients immunodéprimés. Nous…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
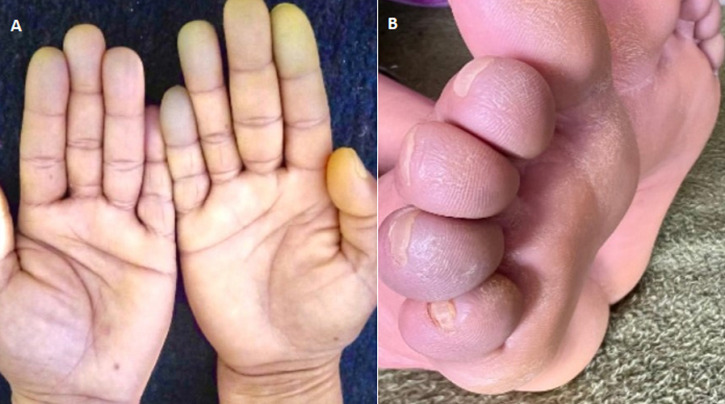 Figure 1
Figure 1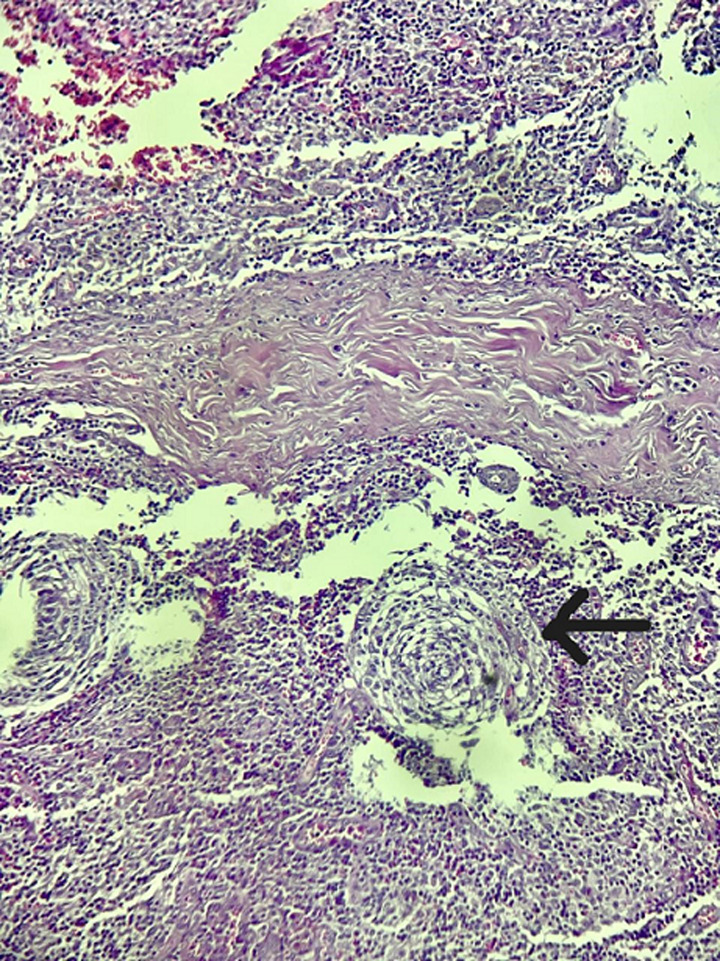 Figure 2
Figure 2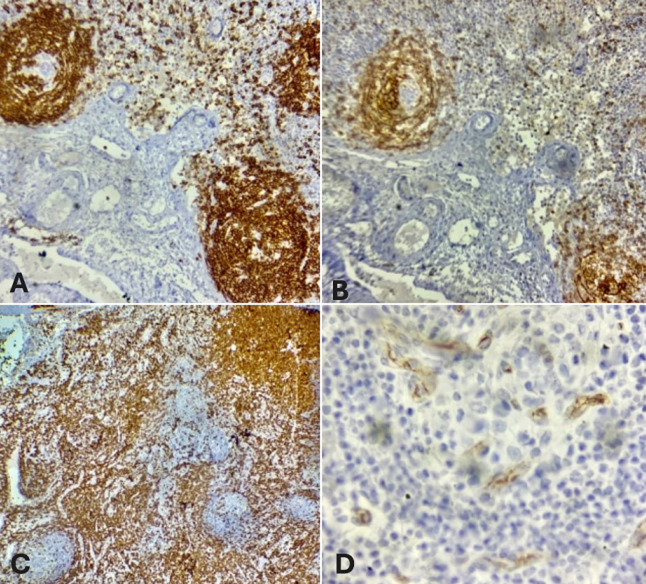 Figure 3
Figure 3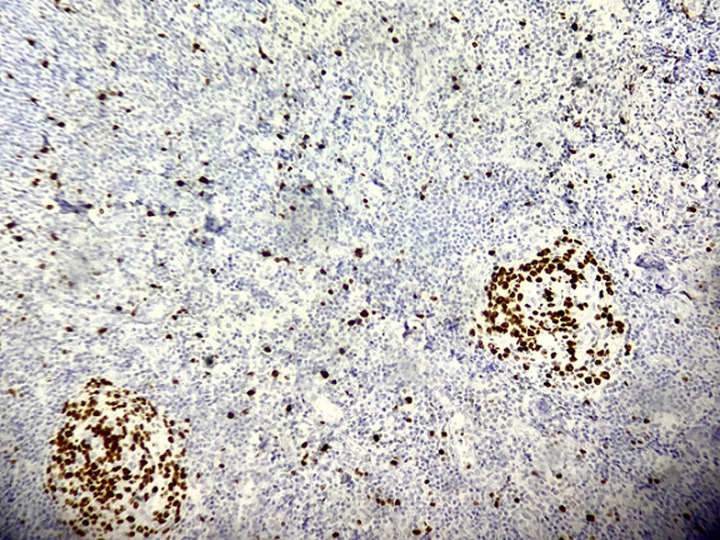 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsViral-associated cancers and disorders · Lymphadenopathy Diagnosis and Analysis · IgG4-Related and Inflammatory Diseases
Introduction
La maladie de Castleman, aussi appelée hyperplasie angiofolliculaire, est une entité rare d'hyperplasie lymphoïde. Elle peut se présenter sous deux formes cliniques principales: la forme unicentrique localisée et la forme multicentrique, systémique. Cette dernière est souvent associée à des symptômes généralisés et à une production excessive de cytokines, notamment IL-6 [1]. Nous présentons ici le cas d'un patient de 65 ans avec une forme multicentrique symptomatique.
Patient et observation
Informations du patient: monsieur MC, âgé de 65 ans, ayant comme antécédents une insuffisance cardiaque chronique traitée par Aldactone® et Lasilix®. Il a consulté initialement au service des urgences pour une douleur abdominale diffuse rebelle au traitement évoluant depuis plusieurs mois. Un scanner abdominal, réalisé dans ce contexte, a permis d'éliminer une urgence chirurgicale et a révélé la présence d'adénopathies mésentériques multiples ainsi qu'une hépato-splénomégalie homogène. L'évolution est marquée par l'apparition d'une polyadénopathie généralisée, associée à des épisodes de syndrome de Raynaud caractérisés par des changements de couleur des doigts et des orteils au froid. Aucune fièvre ni de sueurs nocturnes n'ont été rapportées.
Résultats cliniques: à l'examen clinique, on notait des adénopathies palpables, fermes, mobiles, indolores, principalement cervicales, axillaires et inguinales, mesurant entre 1 et 2 cm, sans signe inflammatoire local. L'examen trouve également des doigts et des orteils gonflés, blancs, cyanosés, froids et insensibles (Figure 1).
A,B) phénomène de Raynaud
Démarche diagnostique: devant le tableau clinique et le contexte épidémiologique, plusieurs diagnostics ont été envisagés initialement: lymphome non hodgkinien, tuberculose, sarcoïdose, infections virales: Epstein-Barr virus (EBV), cytomegalovirus (CMV), Human Immunodeficiency Virus (VIH) et la maladie auto-immune (lupus, syndrome lymphoprolifératif).
Le bilan biologique complet incluant ainsi les sérologies (notamment celles de HHV-8, VIH, CMV et EBV) et l'exploration immunologique s'est révélé sans anomalie significative en dehors d'une élévation de la C-reactive protein (CRP) et d’une anémie inflammatoire. Le scanner cervico-thoraco-abdomino-pelvien a montré des adénopathies multiples: cervicales, axillaires, médiastinales, abdominales et inguinales, associées à une hépatosplénomégalie homogène. Une cervicotomie exploratrice a été réalisée. L'analyse histologique de l'adénopathie réséquée a mis en évidence une hyperplasie folliculaire et vasculaire avec un aspect caractéristique en bulbe d'oignon (Figure 2). L'étude immunohistochimique a montré une positivité pour le cluster de différenciation 3, 20, 23 (CD3, CD20, CD23) et le terminal désoxynucléotidyl transférase (TDT) (Figure 3), avec un index de prolifération Ki-67 élevé (Figure 4). L'ensemble des données cliniques, radiologiques et histologiques a permis de poser le diagnostic de maladie de Castleman multicentrique idiopathique à HHV-8 négatif.
parenchyme ganglionnaire avec une zone de manteau épaissie et des lymphocytes disposés en couches donnant l'aspect en bulbe d'oignon (G x 20)
images microscopiques après marquage immunohistochimique: A) marquage intense des lymphocytes B folliculaires pour le CD20; B) marquage intense des cellules dendritiques folliculaires péri vasculaires pour le CD23; C) marquage intense du fond tumoral riche en lymphocytes T par le CD3; D) marquage de cellules tumorales TDT positives autour des zones en bulbe d'oignon
image microscopique après marquage immunohistochimique par Ki-67 montrant un index de prolifération élevé au sein des centres germinatifs
Intervention thérapeutique: le patient a été orienté vers le service de médecine interne pour une prise en charge thérapeutique spécialisée, où un traitement par tocilizumab 8 mg/kg toutes les 2 semaines a été initié, conformément aux recommandations actuelles pour la forme idiopathique multicentrique de la maladie de Castleman. La tolérance et la réponse clinique ont été surveillées régulièrement par un suivi biologique et radiologique.
Suivi et résultats: suivi en cours: bonne observance et tolérance du traitement, amélioration partielle des symptômes rapportés, pas de complications immédiates notées. La CRP montre une cinétique de baisse progressive.
Consentement éclairé du patient: le patient a donné son consentement oralement pour la publication de ce cas.
Discussion
La maladie de Castleman est une pathologie rare dont l'origine exacte n'est pas entièrement élucidée [1], décrite initialement par Castleman et al. dans les années 1950 [2]. On distingue deux formes cliniques principales: unicentrique, limitée à un seul ganglion ou à une seule région ganglionnaire, et multicentrique, plus rare, impliquant plusieurs ganglions et pouvant s'accompagner de manifestations systémiques. Elle peut être associée à l'infection par le virus HHV-8 [3], notamment chez les patients immunodéprimés, ou être de forme idiopathique [4]. Les manifestations systémiques au cours de cette maladie peuvent être expliquées par la production excessive de cytokines, notamment l'interleukine-6. Ce qui peut contribuer à divers phénomènes paranéoplasiques et à une dérégulation du système immunitaire avec une dysfonction endothéliale menant à des spasmes vasculaires qui se manifestent par le syndrome de Raynaud [5,6].
Le diagnostic de la maladie de Castleman repose principalement sur l'analyse histologique des ganglions lymphatiques. Les éléments caractéristiques incluent une hyperplasie folliculaire, une vascularisation proliférante ainsi qu'un aspect en “bulbe d'oignon” dans les centres germinatifs, notamment dans les formes hyalino-vasculaires [2,5]. L'immunohistochimie est indispensable pour confirmer le diagnostic, montrant typiquement une positivité des marqueurs lymphocytaires: CD3 (cellules T), CD20 (cellules B), CD23 (centres germinatifs), un index Ki-67 élevé, traduisant une forte prolifération cellulaire, et une positivité du CD34 témoignant d'une néoangiogenèse importante. Il est crucial d'identifier la forme exacte de la maladie, car la prise en charge thérapeutique et le pronostic diffèrent radicalement [7].
Le traitement de la forme multicentrique dépend de l'étiologie et de la sévérité clinique, et il est basé sur le rituximab en première ligne pour les formes associées au HHV-8 [4,6]. Seuls ou en association avec une chimiothérapie, les antagonistes de l'IL-6 sont le traitement de choix pour les formes actives idiopathiques comme le siltuximab et le tocilizumab [6,7], et dans les formes sévères ou réfractaires, une corticothérapie, des immunosuppresseurs ou une chimiothérapie peuvent être utilisés, avec un suivi long rigoureux pour surveiller les récidives. Par contre, la forme unicentrique est généralement bénigne et peut être traitée efficacement par l'exérèse chirurgicale complète du ganglion atteint, avec un excellent pronostic [3,8].
Conclusion
La maladie de Castleman, bien que rare, doit être envisagée dans le diagnostic différentiel des adénopathies inexpliquées, notamment lorsqu'elles sont volumineuses, persistantes et atypiques. Le diagnostic repose sur l'histologie. Le traitement repose principalement sur la chirurgie dans les formes unifocales, avec un excellent pronostic en l'absence de signes systémiques. Toutefois, les formes multicentriques nécessitent une prise en charge multidisciplinaire et individualisée. Ce cas souligne l'importance de la collaboration entre cliniciens et anatomopathologistes afin d'optimiser la prise en charge de cette pathologie rare.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Fajgenbaum DC Shilling D Castleman Disease Pathogenesis Hematol Oncol Clin North Am 2018 Feb 32111212915761310.1016/j.hoc.2017.09.002 · doi ↗ · pubmed ↗
- 2Castleman B Towne VW Case records of the Massachusetts General Hospital: Case No. 4023.1N Engl J Med 1954 Jun 1025023100151316594410.1056/NEJM 195406102502308 · doi ↗ · pubmed ↗
- 3Oksenhendler E Boulanger E Galicier L Du MQ Dupin N Martin Aet al High incidence of Kaposi sarcoma-associated herpesvirus-related non-Hodgkin lymphoma in HIV-infected patients with multicentric Castleman disease Blood 2002 Apr 1997233161189576410.1182/blood.v 99.7.2331 · doi ↗ · pubmed ↗
- 4van Rhee F Oksenhendler E Srkalovic G Voorhees PM Lim MS Leblanc Ret al International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease Blood Adv 2020 Dec 8423603960503328494610.1182/bloodadvances.2020003334 PMC 7724917 · doi ↗ · pubmed ↗
- 5Yoshizaki K Matsuda T Nishimoto N Kuritani T Taeho L Aozasa Ket al Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman's disease Blood 1989 Sep 744136072788466 · pubmed ↗
- 6van Rhee F Voorhees PM Dispenzieri A FossåA Srkalovic G Ide Met al International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease Blood 2018 Nov 1513220211521243018117210.1182/blood-2018-07-862334 PMC 6238190 · doi ↗ · pubmed ↗
- 7El-Osta H Kurzrock R Castleman's disease: from basic mechanisms to molecular therapeutics Oncologist 20111644975112144129810.1634/theoncologist.2010-0212 PMC 3228122 · doi ↗ · pubmed ↗
- 8Fajgenbaum DC Uldrick TS Bagg A Frank D Wu D Srkalovic Get al International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease Blood 2017 Mar 2312912164616572808754010.1182/blood-2016-10-746933 PMC 5364342 · doi ↗ · pubmed ↗