Multimodality Imaging in Cardiac Amyloidosis
Mayuresh Chaudhari, Mahi Lakshmi Ashwath

TL;DR
This paper reviews how combining different imaging techniques improves the diagnosis and treatment of heart disease caused by amyloid deposits.
Contribution
The paper emphasizes the integration of multimodal imaging for better diagnosis and management of cardiac amyloidosis.
Findings
Multimodal imaging improves diagnostic accuracy in cardiac amyloidosis.
Each imaging modality has unique strengths in detecting and monitoring amyloidosis.
Integrated imaging guides prognosis and treatment decisions for AL and ATTR amyloidosis.
Abstract
Cardiac amyloidosis is an underdiagnosed cause of heart failure characterized by extracellular deposition of misfolded proteins. Advances in non-invasive imaging, including echocardiography, cardiac magnetic resonance imaging (CMR), and radionuclide imaging, have significantly enhanced the diagnostic accuracy and monitoring of cardiac amyloidosis. This review explores the role of each modality, their individual strengths, and current consensus recommendations. Emphasis is placed on the integration of multimodal imaging to guide diagnosis, prognosis, and therapeutic decisions in both AL and ATTR amyloidosis.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
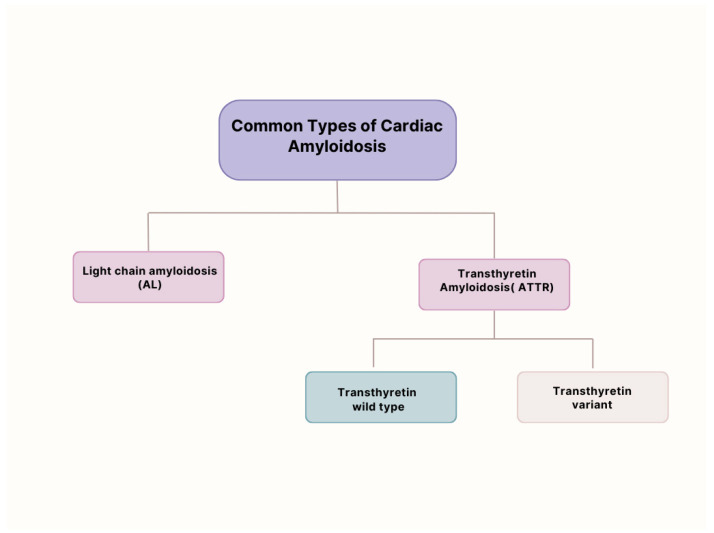 Figure 1
Figure 1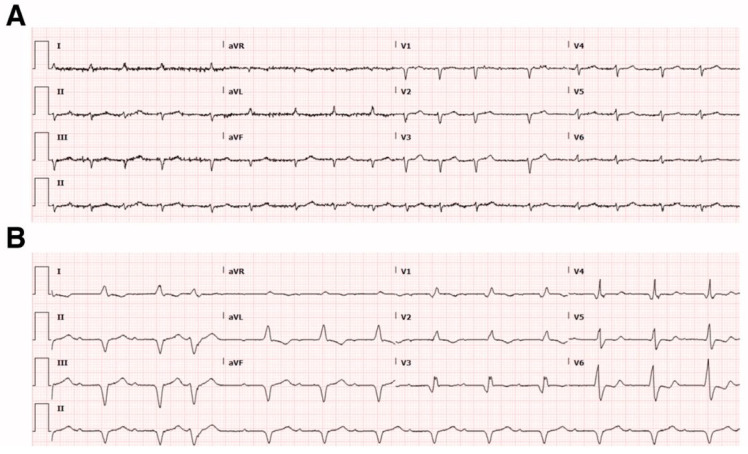 Figure 2
Figure 2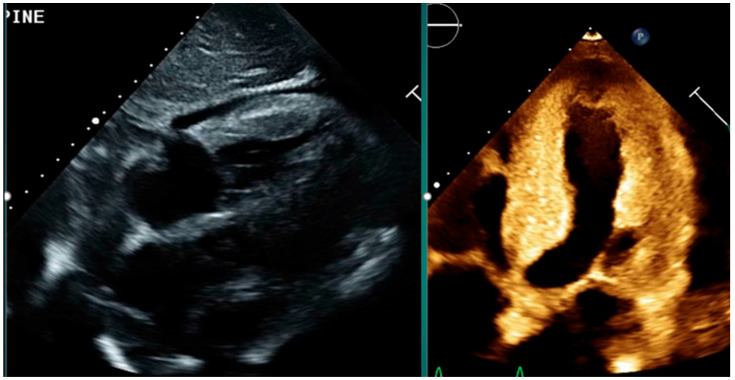 Figure 3
Figure 3 Figure 4
Figure 4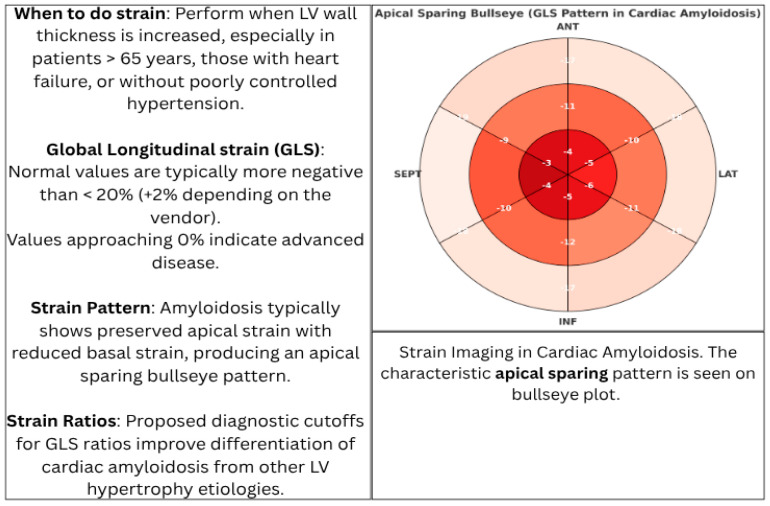 Figure 5
Figure 5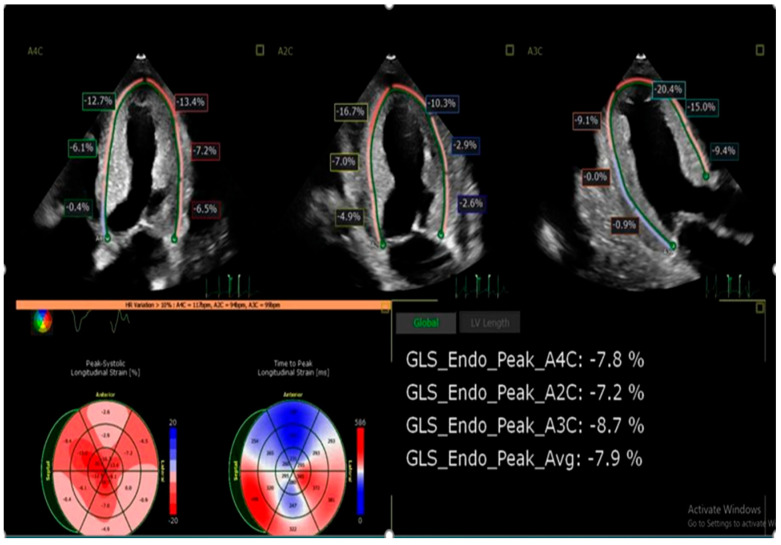 Figure 6
Figure 6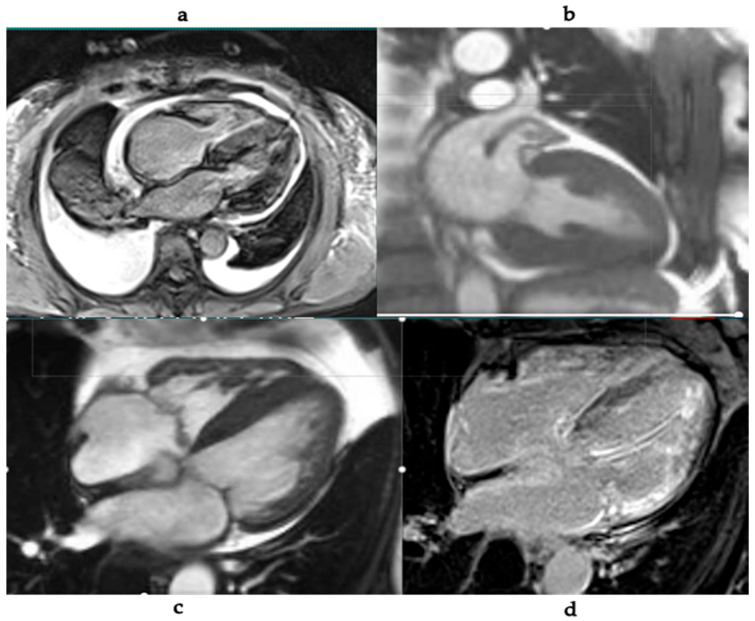 Figure 7
Figure 7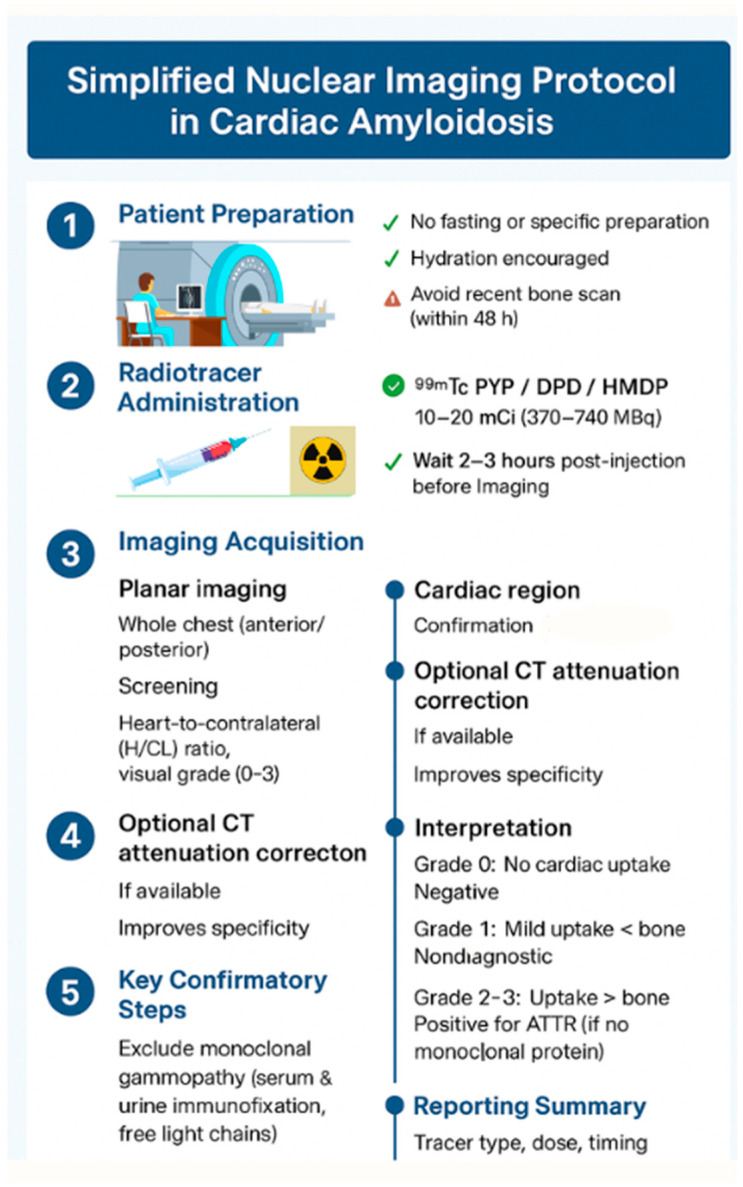 Figure 8
Figure 8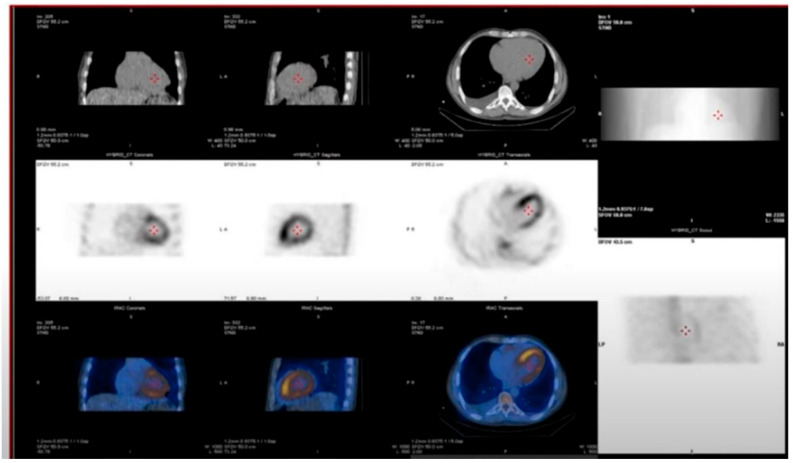 Figure 9
Figure 9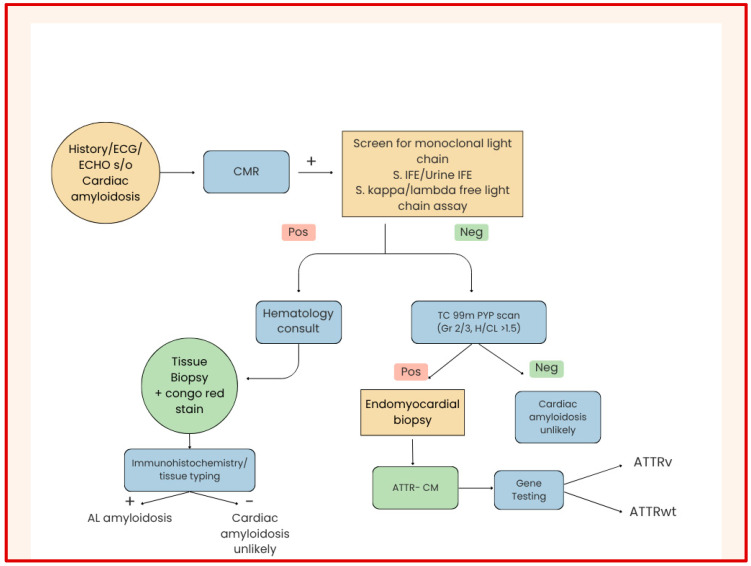 Figure 10
Figure 10Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyloidosis: Diagnosis, Treatment, Outcomes · Pericarditis and Cardiac Tamponade · Cardiac Imaging and Diagnostics
1. Introduction
Cardiac amyloidosis is a progressive and frequently underrecognized cause of heart failure, resulting from the deposition of misfolded protein fibrils within the myocardium [1,2]. Once considered a rare diagnosis, advances in non-invasive imaging and improved clinical awareness have led to increased recognition of this condition, particularly in patients with heart failure with preserved ejection fraction (HFpEF) [3,4]. Over the past decade, cardiac amyloidosis has shifted from an uncommon and often overlooked diagnosis to a condition increasingly identified across diverse cardiovascular populations, driven by the growing understanding that transthyretin amyloidosis is far more prevalent—especially among older adults—and that both AL and ATTR forms frequently mimic more common cardiac diseases. The availability of sensitive monoclonal protein assays, emerging targeted therapies, and the recognition of early extramyocardial manifestations have reinforced the urgent need for timely detection and accurate subtype classification. The two primary forms of cardiac amyloidosis are light-chain (AL) amyloidosis, caused by plasma cell dyscrasias, and transthyretin (ATTR) amyloidosis, which occurs in either a hereditary (ATTRv) or wild-type (ATTRwt) form [5,6]. Early and accurate diagnosis is crucial, as treatment options and prognoses vary significantly between subtypes [7]. Historically, diagnosis relied heavily on tissue biopsy and invasive methods; however, the emergence of advanced cardiac imaging modalities—namely echocardiography, cardiac magnetic resonance imaging (CMR), and radionuclide imaging using bone-avid tracers—has revolutionized the diagnostic approach [8,9,10]. These tools now enable non-invasive detection, precise classification, and longitudinal monitoring of cardiac involvement, while also supporting early identification when therapeutic interventions are most effective. This review highlights the pathophysiology of cardiac amyloidosis and evaluates the role of each imaging modality in diagnosis, risk stratification, and disease monitoring. A multimodality approach, integrating findings from echocardiography, CMR, and nuclear imaging, is increasingly regarded as the gold standard for comprehensive assessment [11,12].
2. Pathophysiology of Cardiac Amyloidosis
Amyloidosis is a disorder characterized by the extracellular deposition of misfolded protein fibrils, which adopt a beta-pleated sheet conformation and disrupt normal tissue architecture and function [13]. In the heart, these deposits increase myocardial stiffness, impair relaxation, and eventually lead to restrictive cardiomyopathy and heart failure [14].
AL amyloidosis results from a clonal proliferation of plasma cells producing excessive light chains (kappa or lambda), which misfold and deposit in various organs including the heart. Cardiac involvement is a major determinant of prognosis in AL amyloidosis, with median survival less than six months in untreated patients presenting with heart failure symptoms [15].
ATTR amyloidosis is caused by the misfolding of transthyretin, a tetrameric protein synthesized primarily by the liver. It transports thyroxine and retinol-binding protein. Two subtypes exist (Figure 1):
- Wild-type ATTR (ATTRwt): Formerly known as senile systemic amyloidosis, this form predominantly affects elderly males and is increasingly recognized as a cause of HFpEF [16,17].
- Variant ATTR (ATTRv): An autosomal dominant condition caused by mutations in the TTR gene, leading to earlier and more aggressive cardiac involvement [18].
Both forms result in progressive amyloid infiltration of the myocardium, leading to diastolic dysfunction, conduction system abnormalities, and arrhythmias [19]. Histologically, the myocardium in amyloidosis demonstrates widespread interstitial amyloid deposits with myocyte atrophy and minimal inflammation. The pattern and extent of amyloid distribution differ between AL and ATTR, influencing imaging characteristics and clinical presentation [20].
Recent studies underscore the need for prompt diagnosis and differentiation between AL and ATTR, as specific therapies—ranging from chemotherapeutic agents to transthyretin stabilizers and gene silencers—are now available, making early identification imperative [21,22].
3. Clinical Presentation and Diagnostic Evaluation
Cardiac amyloidosis commonly presents with nonspecific symptoms, making clinical diagnosis challenging without imaging. Typical manifestations include progressive dyspnea, fatigue, peripheral edema, and signs of heart failure, especially with preserved ejection fraction [23,24]. Additional findings such as orthostatic hypotension, syncope, and arrhythmias are also common [25]. On physical examination, elevated jugular venous pressure, hepatomegaly, and peripheral edema may be observed. Carpal tunnel syndrome, biceps tendon rupture, and lumbar spinal stenosis are clinical red flags that may suggest ATTR amyloidosis [26,27] (Table 1).
Electrocardiographic findings include low QRS voltage (limb leads < 5 mm, precordial leads < 10 mm) and a pseudo-infarct pattern, characterized by Q waves in the absence of coronary artery disease [28,29]. These findings, when paired with imaging evidence of left ventricular hypertrophy, raise suspicion for an infiltrative process [30] (Figure 2).
Routine laboratory tests may reveal elevated cardiac biomarkers such as NT-pro BNP and troponins, which are disproportionately elevated compared to the degree of left ventricular systolic dysfunction [31,32]. Serum and urine protein electrophoresis with immunofixation and serum free light-chain assay are essential to evaluate for AL amyloidosis [33].
Given the nonspecific presentation and serious prognostic implications, imaging plays a pivotal role in raising clinical suspicion, prompting further workup, and guiding diagnosis [34,35].
Imaging Testing
Echocardiogram.Most readily available.Cheapest option.Essentially no contraindications.Almost always the first imaging modality to be done, which helps (1) confirm the clinical suspicion of cardiac dysfunction; (2) raise the possibility of diagnosis of cardiac amyloidosis, prompting further workup; (3) provide longitudinal data on cardiac function; and (4) identify complications.Cardiac MRI.Nuclear imaging.
A study in 2019 by Gilstrap et al. demonstrated the increasing incidence and prevalence of cardiac amyloidosis in men, the elderly, and Black patients, suggesting improved awareness and diagnostic testing through extensive noninvasive imaging [36]. Cardiac amyloidosis should therefore be considered during the initial evaluation of patients > 65 years old hospitalized with heart failure [36].
A multicenter study in 2021 by AbouEzzeddine et al. highlighted the increased prevalence of Transthyretin Amyloid Cardiomyopathy ATTR-CM in a community cohort, with a 2.5% prevalence rate in males and 0% in females [37]. Prevalence increased with age, from 0% in patients aged 60–69 years to 21% in those aged ≥ 90 years (p < 0.001). After adjusting for age, ATTR-CM prevalence differed by sex, with a 10.1% rate in men versus 2.2% in women. ATTR-CM was found in a substantial proportion of HFpEF patients with ventricular wall thickening, particularly in older men. These results suggest that systematic evaluation may substantially increase ATTR-CM diagnosis, enabling therapeutically relevant phenotyping of HFpEF [37].
In a study evaluating outcomes of 1230 patients with cardiomyopathy, during a mean follow-up of 4.4 years, Felker et al. (2000) showed that survival was significantly worse among patients with cardiomyopathy due to infiltrative myocardial disease, including cardiac amyloidosis (adjusted hazard ratio, 4.40; 95% confidence interval, 3.04–6.39) [38].
4. Echocardiography in Cardiac Amyloidosis
Echocardiography is typically the first-line imaging modality in suspected cardiac amyloidosis due to its accessibility, cost-effectiveness, and ability to provide critical functional and structural information. Consensus guidelines from major societies [American Society of Nuclear Cardiology(ASNC), American Heart Association (AHA), American Society of Echocardiography (ASE), Heart Failure Society of America (HFSA), Society for Cardiovascular Magnetic Resonance (SCMR)] support the role of echocardiography as a foundational tool in the diagnostic algorithm for cardiac amyloidosis [39].
Key echocardiographic findings include:
- Increased left ventricular (LV) wall thickness, often concentric and symmetric.
- Sparkling or granular myocardial texture due to amyloid infiltration.
- Biatrial enlargement and dysfunction.
- Thickened valves and interatrial septum.
- Small pericardial effusions [40] (Figure 3).
Tissue Doppler Imaging (TDI) and strain imaging are especially valuable in early disease detection. A hallmark feature is “apical sparing” of longitudinal strain, also described as the “cherry-on-top” pattern. This finding has demonstrated high sensitivity (93%) and specificity (82%) in identifying cardiac amyloidosis and is an accurate, reproducible method of differentiating cardiac amyloidosis from other causes of LV hypertrophy [41] (Figure 4 and Figure 5).
Diastolic dysfunction is universal, often progressing from grade I to restrictive filling patterns [42]. Left atrial dysfunction, independent of cavity size, has also been identified as a predictor for atrial arrhythmias [43,44]. Advanced techniques such as myocardial strain analysis and three-dimensional echocardiography are increasingly utilized for risk stratification and monitoring treatment response [45] (Figure 6).
The relationship between ejection fraction (EF) and global longitudinal strain (GLS) in amyloidosis was evaluated in a 2016 study by Pagourelias et al. They proposed the Ejection fraction strain ratio (EFSR = EF/|GLS|) as a novel parameter [46]. This ratio was significantly higher in CA patients compared to controls, with an EFSR cutoff of 4.1 showing strong differentiating capacity for amyloidosis within LV hypertrophy pathologies [46].
Another study assessed the relative regional strain ratio (RRSR), defined as apical LS divided by mid + basal LS. Patients with low EF and high RRSR (>1.19) had the worst prognosis. This tool is both diagnostic and prognostic, with implications for treatment planning [47].
Among patients with AL, standard echocardiographic measures often remain unchanged at 1 year following chemotherapy, despite reductions in cardiac biomarkers. However, longitudinal strain (LS) is a sensitive marker of pre-treatment cardiac dysfunction, predicts survival beyond biomarkers, and detects early improvement following chemotherapy [48].
Left atrial (LA) structure and function are also impaired in CA. A study evaluating LA strain and strain rate demonstrated significant reductions across all phases (reservoir, conduit, active function) compared to matched controls, independent of LA size, EF, and LV filling pressures [49].
Similarly, in hereditary ATTR amyloidosis, LA function is abnormal irrespective of cavity size, and reduced LA myocardial strain rate during atrial systole is a strong predictor of atrial arrhythmias [50].
Finally, the prevalence and prognostic impact of aortic stenosis (AS) in CA have been evaluated. Among patients with CA, ATTRwt was associated with a higher prevalence of AS compared to ATTRv or AL. However, moderate or greater AS was not associated with worsened outcomes in ATTRwt patients [51].
5. Cardiac MRI in Cardiac Amyloidosis
Cardiac magnetic resonance imaging (CMR) has emerged as a powerful tool in the evaluation of cardiac amyloidosis due to its superior tissue characterization and ability to detect myocardial infiltration. It is particularly useful in differentiating cardiac amyloidosis from other causes of left ventricular hypertrophy [52].
Key CMR findings include:
- Increased LV wall thickness and biatrial enlargement.
- Abnormal myocardial nulling pattern on late gadolinium enhancement (LGE).
- Diffuse subendocardial or transmural LGE patterns.
- Elevated native T1 values and increased extracellular volume (ECV) fraction.
- Presence of intracardiac thrombi and small pericardial effusions [53,54] (Figure 7).
LGE imaging is central to diagnosis, with typical findings including global subendocardial or transmural enhancement. Studies have shown that the extent of enhancement correlates with disease severity and prognosis [55]. Native T1 and ECV mapping further aid in quantifying myocardial involvement and tracking response to therapy [56,57]. CMR also offers excellent reproducibility for assessing cardiac morphology and function, enabling longitudinal monitoring. It is considered particularly helpful when echocardiographic findings are inconclusive [58].
In a 2015 study, the authors demonstrated that in a large cohort of AL and ATTR amyloidosis, patients with positive LGE had worse prognosis than those without [55]. Transmural LGE was associated with the greatest amyloid burden and the poorest survival, independent of other risk factors. Phase-sensitive inversion recovery (PSIR) imaging resolved nulling issues and improved diagnostic accuracy. Importantly, three LGE patterns were described: no LGE, subendocardial LGE, and transmural LGE, with prognosis worsening across this spectrum [55].
Parametric mapping with CMR now permits visualization and quantification of changes in myocardial composition based on T1, T2, T2*, and ECV relaxation times [59]. These techniques improve diagnostic precision, inter-patient comparability, and therapeutic monitoring.
CMR with LGE has demonstrated characteristic subendocardial “tramline” enhancement, which may progress to transmural LV and RV involvement in advanced stages [60]. However, atypical LGE distributions are reported, and renal impairment in amyloidosis often limits gadolinium use [60,61,62]. Native T1 mapping offers a promising non-contrast alternative, though reference ranges in chronic kidney disease require further validation [60,61,62].
ECV quantification serves as a surrogate marker of amyloid burden and carries independent prognostic value [63]. Both native T1 and ECV are elevated in early disease, often before LGE becomes apparent, highlighting their value in early detection [63]. ATTR amyloidosis typically demonstrates higher ECV, while AL tends to have higher native T1 values [64].
In a 2017 consensus statement, Messroghli et al. emphasized the value of standardized mapping techniques for inter-center reproducibility [65]. More recently, Martinez-Naharro et al. (2022) showed that changes in ECV track amyloid burden and correlate independently with prognosis after adjusting for other predictors [66]. In a subsequent study, the same group reported that serial native T1 mapping can monitor treatment response without contrast use, with reduced scan time and improved workflow efficiency [67].
Thus, beyond diagnosis, ECV and T1 mapping provide biomarkers for longitudinal assessment of therapy, prognostication, and monitoring of treatment efficacy [66,67].
6. Nuclear Imaging in Cardiac Amyloidosis
Radionuclide imaging plays a pivotal role in diagnosing transthyretin (ATTR) amyloidosis, particularly in distinguishing it from AL amyloidosis without the need for biopsy [68]. Consensus guidelines from professional societies, including the American Society of Nuclear Cardiology (ASNC), endorse its central role in the noninvasive diagnostic algorithm for ATTR-CM [69].
Imaging Modalities include:
- Planar scintigraphy and SPECT using bone-seeking tracers such as ^99m^Tc-pyrophosphate (PYP), 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD), or hydroxymethylene diphosphonate (HMDP).
- SPECT/CT, which provides improved anatomical localization and helps distinguish blood pool activity from true myocardial uptake.
- PET imaging with amyloid-specific tracers (e.g., ^18^F-florbetapir, ^18^F-florbetaben, ^11^C-PiB), which allow high-resolution molecular imaging of amyloid deposits [70,71] (Figure 8).
Planar imaging is simple and widely available. Myocardial uptake is quantified by calculating the heart-to-contralateral chest (H/CL) ratio, with values > 1.5 suggestive of ATTR amyloidosis [72]. Visual grading against rib uptake is also used. Whole-body planar imaging may provide adjunctive evidence of systemic amyloid burden, such as tracer uptake in the shoulder or hip girdles, a specific feature of systemic ATTR amyloidosis [73].
SPECT and SPECT/CT add three-dimensional detail, enabling separation of blood pool activity from myocardial signal and better visualization of uptake in the interventricular septum, which is commonly involved in amyloidosis [74] (Figure 9).
Bone scintigraphy with tracers such as PYP, DPD, and HMDP has shown high specificity and positive predictive value for ATTR amyloidosis when combined with negative monoclonal protein studies [75]. This approach now enables non-biopsy diagnosis of ATTR-CM in the majority of patients [76].
PET amyloid imaging, although less widely available, offers superior spatial resolution and quantification of amyloid burden. ^18F-florbetapir has demonstrated utility in detecting cardiac amyloid deposits even at early stages, particularly in AL amyloidosis [77]. ^11^C-PiB PET has also successfully visualized cardiac amyloid, showing greater uptake in AL compared to ATTR amyloidosis [78].
The adoption of bone-avid tracer scintigraphy for ATTR-CM has grown substantially over the past decade. Successive ASNC surveys have shown increased utilization, broader adoption by clinicians across specialties, and evolving imaging protocols [79].
Thus, radionuclide imaging is now firmly established as a cornerstone modality in the diagnosis and risk stratification of cardiac amyloidosis, with bone scintigraphy enabling noninvasive diagnosis of ATTR-CM and PET imaging providing complementary insights into amyloid biology.
7. Current Guidelines and Consensus Recommendations
The increasing availability and accuracy of multimodality imaging techniques have been incorporated into contemporary guidelines for the diagnosis of cardiac amyloidosis. Several professional societies have issued expert consensus statements to standardize the diagnostic approach [80].
The 2021 international expert consensus document by ASNC, AHA, ASE, EANM, HFSA, ISA, SCMR, and SNMMI recommends the following [81]:
- Initial Screening: Begin with clinical suspicion based on symptoms and red flags; perform serum/urine protein electrophoresis and free light-chain assay.
- Echocardiography: As the first-line imaging modality, assess for structural and functional abnormalities, including apical sparing on strain imaging.
- CMR: Recommended when echocardiographic findings are inconclusive or when more detailed tissue characterization is needed.
- Radionuclide Imaging: ^99m^Tc PYP, DPD, or HMDP scintigraphy is recommended for patients without evidence of monoclonal gammopathy to confirm ATTR amyloidosis (Figure 10 and Table 2).
If bone scintigraphy demonstrates Grade 2 or 3 myocardial uptake with no evidence of plasma cell dyscrasia, a non-biopsy diagnosis of ATTR amyloidosis can be established [82].
These guidelines promote a non-invasive, algorithmic approach, reducing the need for endomyocardial biopsy in most patients and improving diagnostic confidence.
The integration of multimodality imaging findings with laboratory and clinical data ensures accurate subtype differentiation and guides appropriate therapy [80,81,82] (Table 3).
8. Practical Considerations, Pitfalls, and Recent Advances
Despite the high diagnostic accuracy of bone-avid radiotracers such as ^99mTc-PYP, DPD, and HMDP for transthyretin cardiac amyloidosis (ATTR-CM), several pitfalls may lead to false-positive or false-negative results. False-positive uptake may occur in conditions associated with myocardial injury, including acute myocardial infarction, myocarditis, pericarditis, or significant left ventricular hypertrophy unrelated to amyloidosis [83]. Blood pool activity—particularly in patients with atrial fibrillation, renal dysfunction, or low cardiac output—can mimic myocardial uptake on planar imaging, underscoring the importance of SPECT confirmation to ensure true myocardial localization [84]. Rib fractures, valve calcification, and overlapping skeletal uptake may also contribute to false-positive results [85]. Importantly, the presence of a monoclonal protein can confound interpretation, as AL amyloidosis occasionally demonstrates low-grade tracer uptake; in such cases, radionuclide scintigraphy cannot reliably differentiate AL from ATTR, and biopsy is required [83].
Conversely, false-negative results may occur in early or minimal amyloid infiltration, particularly in certain hereditary ATTR variants with low tracer affinity [86]. Patients with AL amyloidosis may also show absent or minimal uptake because bone tracers exhibit much weaker binding to AL fibrils [83]. Technical limitations—including inadequate imaging delay (e.g., imaging before sufficient blood pool clearance), improper region-of-interest selection, or attenuation artifacts—can further reduce sensitivity [84,87]. In rare cases of advanced diffuse infiltration, near-equal myocardial and skeletal uptake may diminish contrast on planar imaging, leading to under-recognition of disease [88].
Given these potential pitfalls, bone scintigraphy results must always be interpreted alongside monoclonal protein testing, echocardiographic or CMR findings, and the overall clinical context to avoid misclassification and ensure accurate amyloid subtype differentiation.
Although advances in multimodality imaging have dramatically reduced the need for tissue biopsy in many patients with suspected cardiac amyloidosis, biopsy remains essential in selected cases. When a patient has strong clinical, laboratory, or imaging features suggestive of amyloidosis, yet radionuclide scintigraphy, echocardiography, or CMR findings are inconclusive or negative, an endomyocardial biopsy should be pursued to avoid missed or delayed diagnosis [89]. This scenario is particularly relevant in AL amyloidosis, where bone scintigraphy may be negative or equivocal due to the lower affinity of bone-avid tracers for AL fibrils [76,83]. Additionally, certain ATTRv mutations and early-stage ATTR disease may exhibit minimal or absent uptake on bone scintigraphy, leading to false-negative noninvasive results [86].
Biopsy is also indicated when a monoclonal gammopathy is present, since noninvasive imaging cannot reliably exclude AL cardiac amyloidosis in this setting. Misclassification of AL as ATTR has major therapeutic consequences, given the urgency of chemotherapy-based treatment for AL amyloidosis. In such cases, either endomyocardial biopsy or biopsy of an involved extracardiac organ (e.g., fat pad, kidney, or bone marrow with mass spectrometry) is critical for definitive amyloid typing [90].
Ultimately, when the pre-test probability of cardiac amyloidosis remains high—based on clinical red flags (e.g., intolerance to heart failure therapy, neuropathy, bilateral carpal tunnel syndrome), biomarker profile, or imaging patterns—histologic confirmation becomes a key step to ensure accurate subtype classification and guide appropriate therapy.
A significant recent advancement in cardiac amyloidosis imaging is the development of 124I-evuzamitide, a pan-amyloid–binding PET tracer designed to detect amyloid deposits across multiple organs. In a phase 1/2 study, 124I-evuzamitide PET/CT demonstrated high sensitivity (≈93.6%) for detecting cardiac amyloid, with uptake observed in both ATTR and AL amyloidosis. Importantly, myocardial tracer uptake correlated with clinical manifestations and cardiac involvement, supporting its potential diagnostic and prognostic utility [91]. In several cases, evuzamitide identified cardiac amyloid deposition even when conventional bone-avid scintigraphy (e.g., ^99mTc-PYP) was negative, suggesting greater sensitivity for early or subtle myocardial infiltration [92].
Based on these promising findings, the U.S. Food and Drug Administration granted Breakthrough Therapy Designation for evuzamitide as a diagnostic imaging agent for suspected cardiac amyloidosis, with additional Orphan Drug Designations from both the FDA and the European Medicines Agency [93]. As a pan-amyloid tracer, evuzamitide enables whole-body visualization of amyloid burden, including cardiac, renal, hepatic, splenic, and soft-tissue involvement, providing a comprehensive assessment of systemic disease [94]. If validated in larger multicenter studies, 124I-evuzamitide may significantly enhance early detection, facilitate disease monitoring, and refine therapeutic decision-making by quantifying global amyloid load.
In a comprehensive State-of-the-Art Review, Rapezzi et al. critically compared contemporary guidance documents from major international societies—including ASNC, AHA, ESC, HFSA, ISA, and SCMR—and highlighted both areas of agreement and important discrepancies within current recommendations for the diagnosis and management of cardiac amyloidosis [95]. The review notes strong consensus across societies regarding the need for heightened clinical awareness, early recognition of red-flag features, and the central role of multimodality imaging—particularly echocardiography, CMR, and bone-avid nuclear scintigraphy—in diagnostic pathways. All societies universally emphasize differentiating AL from ATTR amyloidosis, reflecting fundamental differences in treatment urgency, therapeutic strategies, and prognosis. However, the authors underscore meaningful variations between guidelines in the sequencing of tests, interpretation of radionuclide scintigraphy (particularly Grade 1 uptake), and thresholds for advancing to endomyocardial biopsy, especially when a monoclonal gammopathy is present [96,97]. Terminology for diagnostic certainty (“suspected,” “probable,” “definite” amyloidosis) and criteria defining myocardial involvement also differ subtly among professional societies, potentially contributing to inconsistent diagnostic approaches and variability in clinical decision-making. Additionally, the review highlights uneven integration of emerging modalities—such as T1/ECV mapping, strain imaging, and amyloid-specific PET tracers—across society documents, despite growing evidence supporting their diagnostic and prognostic value [98]. Rapezzi and colleagues argue that these discrepancies may delay diagnosis, increase misclassification risk, and complicate implementation of evolving therapeutic strategies, particularly in regions with differing imaging availability. Accordingly, the authors call for greater international harmonization, proposing that future guidelines adopt unified terminology, standardized imaging criteria, and algorithmic pathways that fully integrate both established and emerging technologies to improve diagnostic precision and patient outcomes [95,96,97,98]. Important key studies in cardiac amyloidosis imaging have been summarized in the reference table below [Table 4].
9. Conclusions
Cardiac amyloidosis, once considered rare, is now increasingly diagnosed due to advances in multimodality imaging and heightened clinical awareness. Differentiating between AL and ATTR subtypes is essential for appropriate therapy, and this is best achieved through an integrated diagnostic strategy.
Echocardiography remains the frontline modality, while cardiac MRI adds vital tissue characterization, and nuclear imaging—especially with bone-avid tracers—has revolutionized non-invasive diagnosis of ATTR. These tools, when used in combination and guided by current consensus recommendations, offer high diagnostic accuracy and enable early therapeutic intervention.
Ongoing advancements in imaging techniques and therapeutic options continue to improve outcomes for patients with cardiac amyloidosis. Multidisciplinary collaboration and adherence to standardized algorithms will be key in optimizing care.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ruberg F.L. Berk J.L. Transthyretin (TTR) cardiac amyloidosis Circulation 20121261286130010.1161/CIRCULATIONAHA.111.07891522949539 PMC 3501197 · doi ↗ · pubmed ↗
- 2Martinez-Naharro A. Hawkins P.N. Fontana M. Cardiac amyloidosis Clin. Med.201818 S 30S 3510.7861/clinmedicine.18-2-s 30PMC 633403529700090 · doi ↗ · pubmed ↗
- 3González-López E. Gallego-Delgado M. Guzzo-Merello G. de Haro-Del Moral F.J. Cobo-Marcos M. Robles C. Bornstein B. Salas C. Lara-Pezzi E. Alonso-Pulpon L. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction Eur. Heart J.2015362585259410.1093/eurheartj/ehv 33826224076 · doi ↗ · pubmed ↗
- 4Mohammed S.F. Mirzoyev S.A. Edwards W.D. Dogan A. Grogan D.R. Dunlay S.M. Roger V.L. Gertz M.A. Dispenzieri A. Zeldenrust S.R. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction JACC Heart Fail.2014211312210.1016/j.jchf.2013.11.00424720917 PMC 3984539 · doi ↗ · pubmed ↗
- 5Merlini G. Bellotti V. Molecular mechanisms of amyloidosis N. Engl. J. Med.200334958359610.1056/NEJ Mra 02314412904524 · doi ↗ · pubmed ↗
- 6Gillmore J.D. Damy T. Fontana M. Hutchinson M. Lachmann H.J. Martinez-Naharro A. Quarta C.C. Rezk T. Whelan C.J. Gonzalez-Lopez E. A new staging system for cardiac transthyretin amyloidosis Eur. Heart J.2018392799280610.1093/eurheartj/ehx 58929048471 · doi ↗ · pubmed ↗
- 7Grogan M. Dispenzieri A. Gertz M.A. Light-chain cardiac amyloidosis: Strategies to promote early diagnosis and cardiac response Heart 20171031065107210.1136/heartjnl-2016-31070428456755 PMC 5566095 · doi ↗ · pubmed ↗
- 8Phelan D. Collier P. Thavendiranathan P. PopovićZ.B. Hanna M. Plana J.C. Marwick T.H. Thomas J.D. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis Heart 2012981442144810.1136/heartjnl-2012-30235322865865 · doi ↗ · pubmed ↗