Aglodorols A–J, undescribed terpenoids with multidimensional neuroprotective activities from Aglaia odorata Lour
Meng Ding, Yue-Han Wang, Chen-Hao Liu, Wang-Xiao Tan, Li-Ming Hu, Ke-Wu Zeng, Peng-Fei Tu, Yong Jiang

TL;DR
This study discovers new terpenoids from Aglaia odorata that show strong neuroprotective effects in various cell models of nerve injury.
Contribution
The paper reports the isolation of unprecedented triterpenoids and diterpenoids with novel multidimensional neuroprotective activities.
Findings
Compounds 3, 9–11, 13, 17, and 18 protected PC12 cells from OGD/R-induced nerve injury at 10 µM.
Triterpenoids 1, 2, and 11 showed strong activity against RSL3-induced PC12 cell death with EC50 values of 1.16–1.74 µM.
Compound 22 exhibited the highest inhibition of NO release in LPS-activated BV2 cells with an IC50 of 22.41 µM.
Abstract
Three unprecedented triterpenoids (1–2, 8), seven novel diterpenoids (13–16, 19–21), and 12 known compounds (3–7, 9–12, 17–18, 22) were isolated from the tender branches and leaves of Aglaia odorata Lour. Structural elucidation was achieved through integrated spectroscopic analysis, quantum chemical calculations (NMR/ECD), and single-crystal X-ray diffraction. All isolates were evaluated for neuroprotective effects. Compounds 3, 9–11, 13, 17, and 18 showed significant protective effects against oxygen-glucose deprivation/reperfusion (OGD/R)-mediated nerve injury in PC12 cells at 10 µM, while compounds 3 and 19 exhibited potent anti-excitotoxicity activity in the L-glutamate-induced HT22 cells at 20 µM. Strikingly, triterpenoids 1, 2, and 11 displayed remarkable activity against RSL3-induced PC12 cell death with EC50 values ranging from 1.16 to 1.74 μM. Compound 22 exhibited the most…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
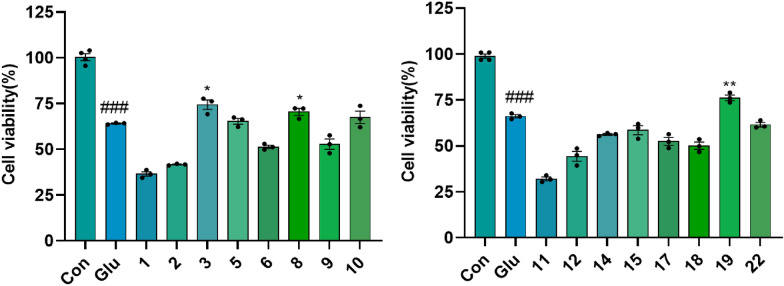 Figure 10
Figure 10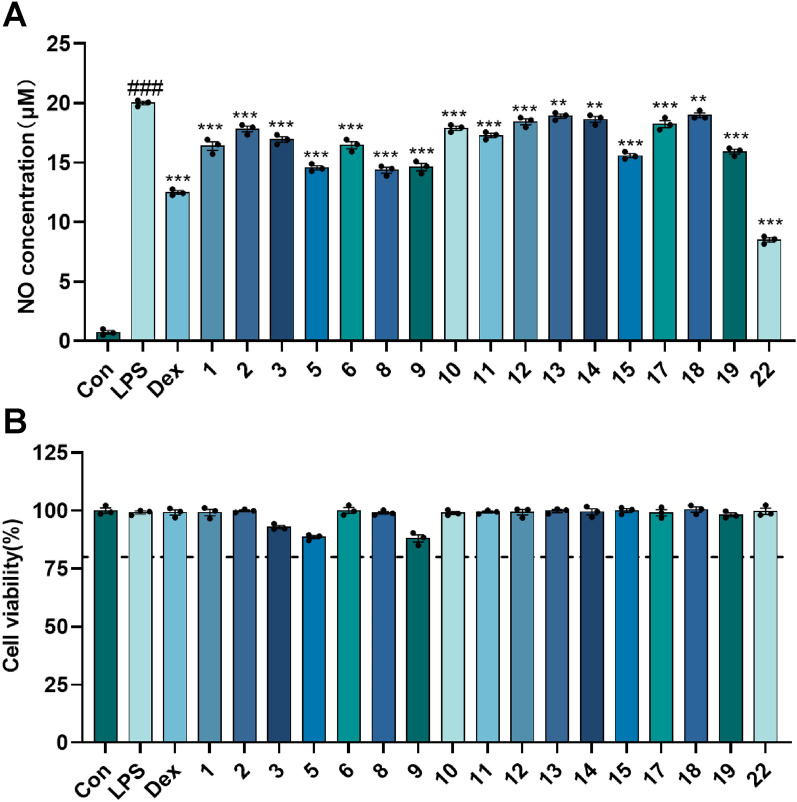 Figure 11
Figure 11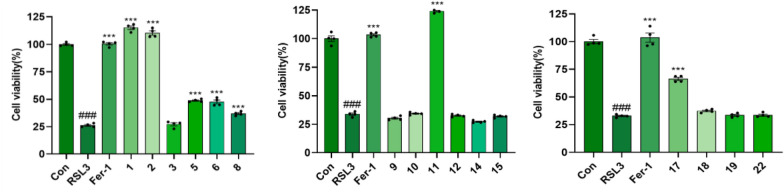 Figure 12
Figure 12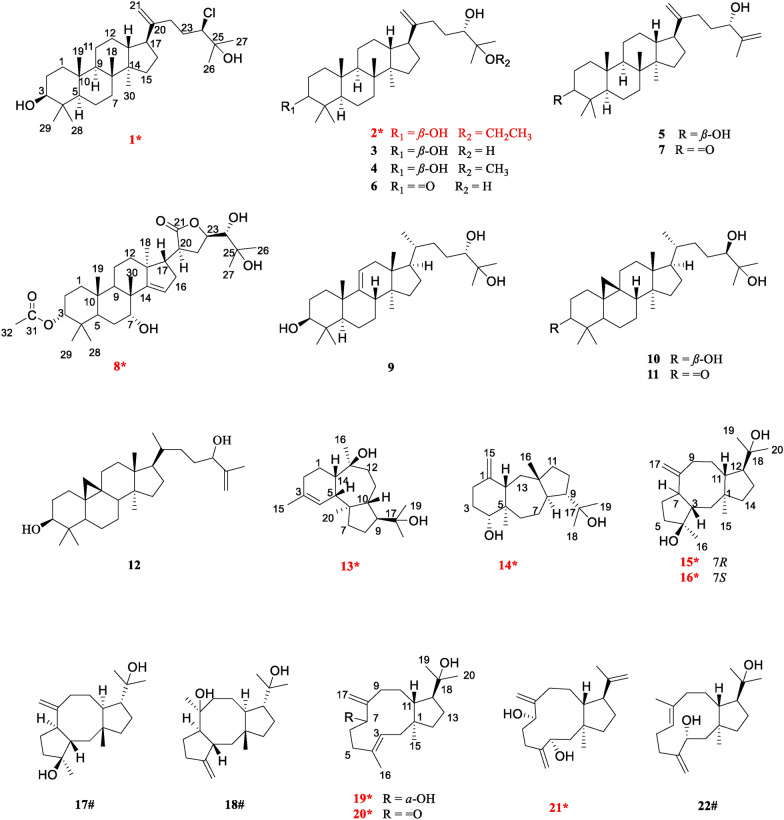 Figure 1
Figure 1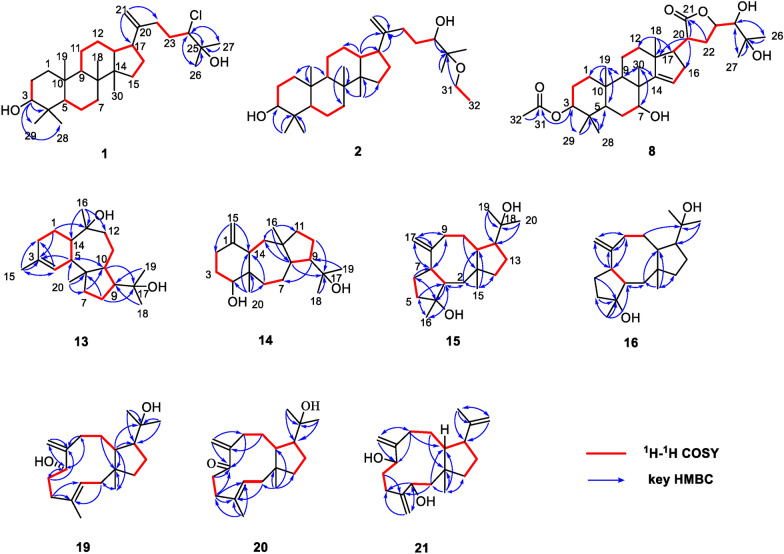 Figure 2
Figure 2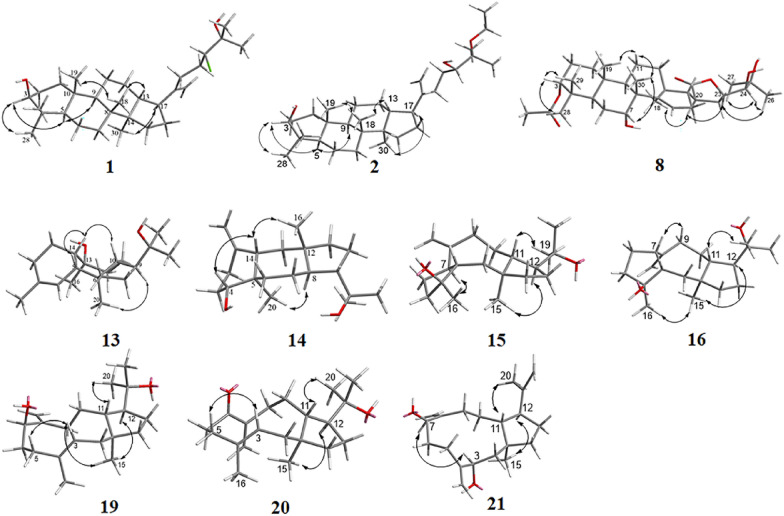 Figure 3
Figure 3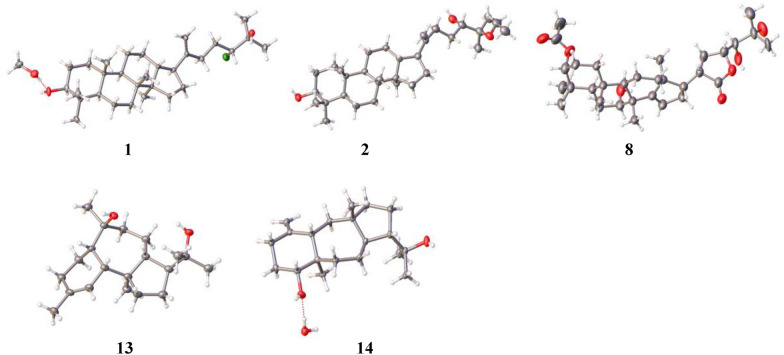 Figure 4
Figure 4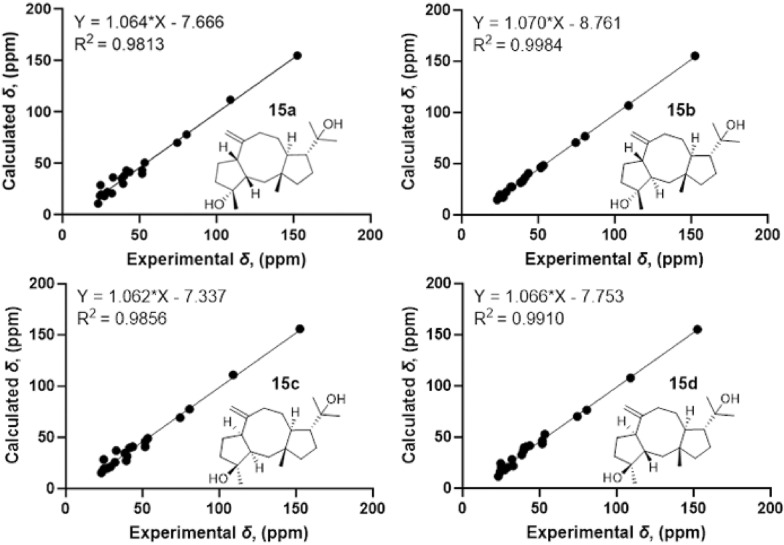 Figure 5
Figure 5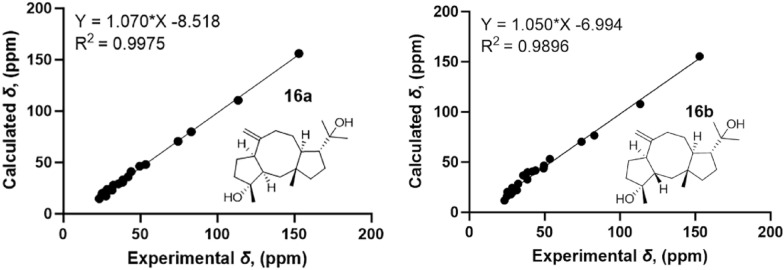 Figure 6
Figure 6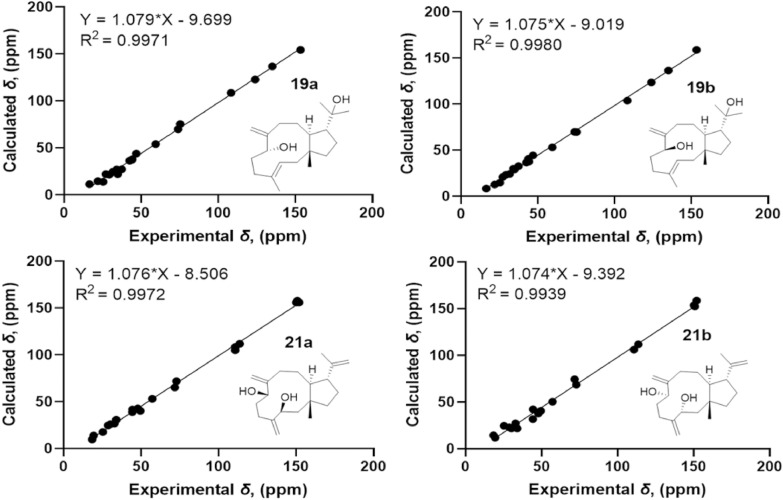 Figure 7
Figure 7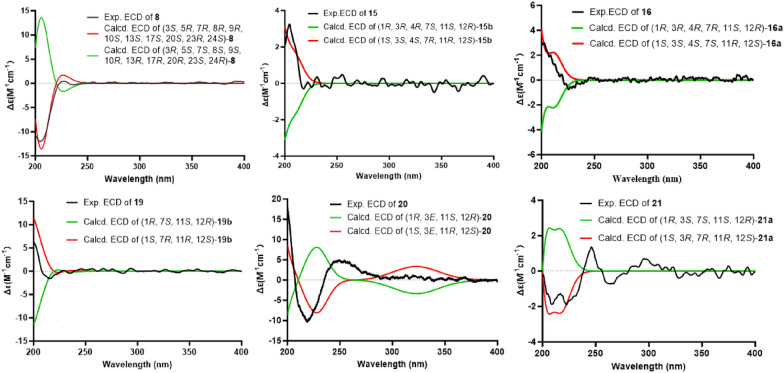 Figure 8
Figure 8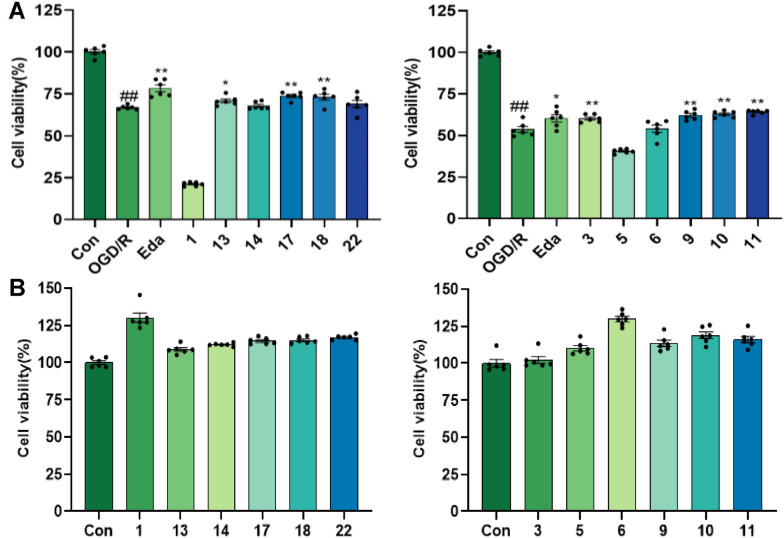 Figure 9
Figure 9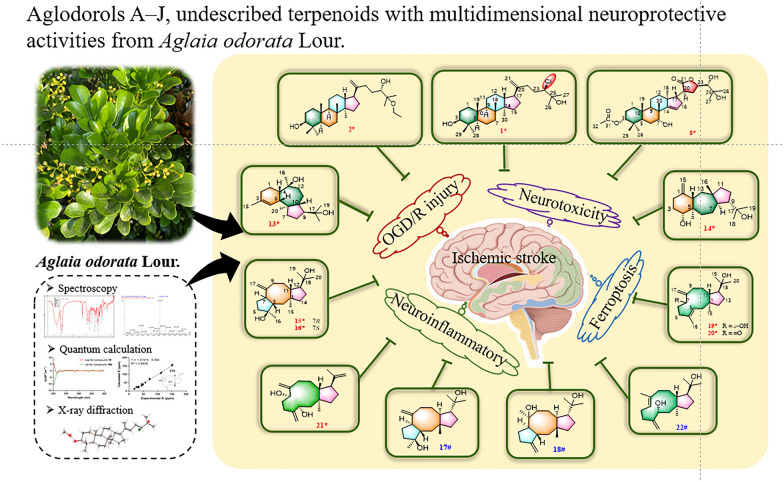 Figure 13
Figure 13- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
- —http://dx.doi.org/10.13039/100014103Key Technology Research and Development Program of Shandong Province
- —http://dx.doi.org/10.13039/501100002858China Postdoctoral Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhytochemical compounds biological activities · Bioactive Compounds in Plants · Biological Activity of Diterpenoids and Biflavonoids
Introduction
The genus Aglaia, the largest genus of the family Meliaceae, contains approximately 150 species and primarily distributed across tropical and subtropical Asia [1, 2]. The genus holds significant agricultural and industrial importance, with selected species serving as regionally valuable fruit crops and premium timber sources [2, 3].
Aglaia odorata Lour., widely planted as a greening tree in China, has been systematically documented in Chinese medical compendia for its properties in activating blood circulation, resolving stasis, subduing swelling, and relieving pain [4]. Additionally, the flowers of Aglaia odorata are brewed into a floral tea, which is traditionally consumed to alleviate alcohol intoxication and cleanse the lungs. Contemporary studies have substantiated its broad bioactivity profile encompassing antiproliferative, antifungal, antiviral, and insecticidal effects [1]. Notably, recent investigations revealed its promising neuroprotective potential. Wang et al. demonstrated the cerebroprotective efficacy of A. odorata extract using both an in vivo middle cerebral artery occlusion model and an in vitro oxygen-glucose deprivation/reperfusion (OGD/R) model [4]. Yin et al. further investigated the anti-neuroinflammatory activity of the extract and isolates of A. odorata using the lipopolysaccharide (LPS)-induced BV-2 cell model and evaluated the therapeutic effect of rocaglaol on neuroinflammation in mice [5].
Studies have shown that the pathophysiological cascade following ischemic stroke involves intricate interplay between neuronal autotoxicity and peripheral damage mechanisms. This complex process includes excitatory amino acid surges, oxidative stress amplification, dysregulated inflammatory responses, and programmed cell death pathways [6]. To adequately investigate the neuroprotective effects of A. odorata, the phytochemical exploration of A. odorata was performed to give the isolation and structural elucidation of 22 terpenoids, including 10 previously unreported compounds aglodorols A–J (1–2, 8, 13–16, 19–21), and 12 known compounds (3–7, 9–12, 17–18, 22). The neuroprotective activities of these isolates were evaluated by a comprehensive multi-model screening system including OGD/R model, the glutamate-induced neurotoxicity model, the LPS-induced neuroinflammation model, and the ferroptosis model.
Results and discussion
Structural elucidation of compounds
A total of 22 secondary metabolites were isolated, including 10 new compounds (1–2, 8, 13–16, and 19–21) along with 12 known compounds (3–7, 9–12, 17–18, and 22) (Fig. 1). Notably, compound 1 is a rare chlorine-containing dammarane-type triterpene, and compound 8 is the first reported apotirucallane-type triterpenoid containing a five-membered lactone ring in the side chain. In addition, dolastane-type diterpenes (13–14) and fusicoccane-type diterpenes (15–18), believed to be found only in sea creatures [7], have now been reported in higher plants for the first time. The remaining 12 known compounds were identified as 3,24,25‐trihydroxy‐dammar‐20‐ene (3) [8], 25-methoxy-5α-dammar-20-en-3β,24-diol (4) [9], dammara-20,25-dien-3β,24α-diol (5) [10], 24,25-dihydroxy-dammar-20-en-3-one (6) [8], 24-hydroxydammara-20,25-dien-3-one (7) [9], (24S)-5α-lanost-9(11)-ene-3β,24,25-triol (9) [11], (24R)-cycloartane-3β,24,25-triol (10) [12], (24R)-9,19-cyclolanost-3-one-24,25-diol (11) [13], 3β-hydroxy-25-methylenecycloartan-24-ol (12) [14], barbifusicoccin A (17) [15], barbifusicoccin B (18) [15], and (1R, 3S, 7E, 11S, 12R)-dolabella-4(16),7-dien-3,18-diol (22) [16] by comparison of spectroscopic data with literature. In addition, the absolute configurations of the known compounds 17, 18, and 22 were determined through ECD calculations.Fig. 1. Structures of compounds 1–22 (new compounds were marked with *, and known compounds featuring first-established stereochemical configurations with #)
Aglodorol A (1) was obtained as colorless massive crystals, mp 167–168 °C, whose molecular formula was deduced to be with a chlorine atom as C_30_H_51_O_2_Cl, from the relative abundance ratio of 3:1 for [M + HCOO]^−^ ion at m/z 523.3557 (calcd for C_31_H_52_O_4_Cl, 523.3560) and m/z 525.3554 [M + HCOO + 2]^−^ in the HRESIMS spectrum. The IR spectrum shows stretching bands of hydroxy group (3320 cm^− 1^), chlorine substitution (806 cm^− 1^), and double bond (1643 cm^− 1^). The UV spectrum exhibits maximum absorption bands at 203 nm, suggesting the presence of double bond.
The ^1^H NMR data present seven methyl singlet [δH 1.31 (H_3_-26), 1.29 (H_3_-27), 0.98 (H_3_-18), 0.97 (H_3_-28), 0.87 (H_3_-30), 0.84 (H_3_-19), and 0.77 (H_3_-29)] and two terminal olefinic methylene [δH 4.78 and 4.70 (s, H_2_-21)] protons (Table 1). The analysis of ^13^C NMR and HSQC spectra of compound 1 reveals 30 carbon signals, including seven methyl, 11 methylene, six methine, and six quaternary carbons, which are very similar to those of the reported compound 3 [8]. Given the molecular formula of 1, it was deduced that one of the three hydroxy groups in 3 is substituted by a chlorine atom. Detailed comparison of the NMR data shows that the most significant difference in chemical shifts between 1 (δC 74.0) and 3 (δC 78.3) is at C-24, suggesting that the chlorine atom may be substituted at C-24. The ^1^H–^1^H COSY correlations (H-22a/H-23a and H-23b/H-24) and the HMBC correlations (from H-24 to C-22/C-23/C-25/C-26/C-27, from H_3_-26 to C-24/C-25/C-27, and from H_3_-27 to C-24/C-25/C-26) determine the structure of the side chains (Fig. 2). Therefore, the planar structure of 1 was established. Considering the relative scarcity of naturally occurring chlorinated phytochemicals, the possibility of 1 being an artifact was rigorously investigated. A freshly prepared methanol extract of the title plant was immediately analyzed using UPLC/Qtrap-MS/MS in multiple reaction monitoring (MRM) mode. The metabolite 1 was unambiguously detected in this initial extract (Fig. S11), which effectively rules out its formation during long-term storage or the subsequent isolation procedures. Thus, we propose 1 as an enzyme-catalyzed halogenated product, with the responsible halogenase originating either from the plant’s own biosynthetic machinery [17] or from endophytic bacteria associated with the plant host [18]. Table 1^1^H (500 MHz) and ^13^C (125 MHz) NMR data of compounds 1, 2, and 8 (δ in ppm, J in Hz) in Chloroform-d_1_No.128δH (J in Hz)δC, typeδH (J in Hz)δC, typeδH (J in Hz)δC, type1α0.95, m39.2, CH_2_0.97, m39.1, CH_2_1.36, m33.3, CH_2_1β1.71, m1.71, m2α1.59, m27.6, CH_2_1.57, m27.4, CH_2_1.59, m22.87, CH_2_2β1.62, m1.62, m1.88, m33.20, dd (11.4, 4.8)79.1, CH3.20, dd (11.5, 4.9)79.0, CH4.65, m78.2, CH439.1, C39.0, C36.2, C50.73, d (11.6)56.0, CH0.73, dd (11.9, 2.2)55.8, CH1.95, dd (11.4, 4.2)42.1, CH6α1.54, m18.4, CH_2_1.53, m18.3, CH_2_1.73, m23.7, CH_2_6β1.42, m1.44, m7α1.57, m35.5, CH_2_1.56, m35.4, CH_2_3.92, t (2.9)72.4, CH7β1.23, m1.28, m840.6, C40.4, C44.6, C91.28, m51.1, CH1.30, m50.9, CH2.00, dd (11.4, 8.2)41.7, CH1037.3, C37.2, C37.7, C11α1.51, m21.5, CH_2_1.51, m21.3, CH_2_1.71, m16.3, CH_2_11β1.19, m1.50, m12α1.05, m25.2, CH_2_1.05, m25.0, CH_2_2.12, m32.2, CH_2_12β1.55, m1.56, m131.69, m45.0, CH1.67, m45.5, CH47.0 C1449.6, C49.4, C161.6, C15α1.11, m31.4, CH_2_1.10, m31.4, CH_2_5.49, d (2.9)119.4, CH15β1.61, m1.60, m16α1.93, m29.0, CH_2_1.90, m29.0, CH_2_2.30, m33.2, CH_2_16β1.39, m1.44, m172.16, m47.4, CH2.20, m48.2, CH2.29, m54.1, CH180.98, s15.8, CH_3_0.97, s15.6, CH_3_1.06, s20.3, CH_3_190.84, s16.4, CH_3_0.84, s16.2, CH_3_0.90, s15.3, CH_3_20151.4, C153.0, C2.76, m39.7, CH21a4.78, s108.2, CH_2_4.71, s107.5, CH_2_177.9, C = O21b4.70, s4.76, s22a2.08, m31.8, CH_2_2.00, m31.2, CH_2_2.24, m29.8, CH_2_22b2.33, m2.30, m2.41, q (11.8)23a1.72, m31.7, CH_2_1.48, m29.8, CH_2_4.62, m77.6, CH_2_23b2.01, m1.52, m243.86, d (11.3)74.0, CH3.46, m76.4, CH3.29, d (1.3)76.1, CH2572.9, C77.3, C72.7, C261.31, s26.7, CH_3_1.11, s19.4, CH_3_1.34, s26.8, CH_3_271.29, s25.2, CH_3_1.14, s21.5, CH_3_1.29, s26.7, CH_3_280.97, s28.2, CH_3_0.98, s28.0, CH_3_0.85, s27.7, CH_3_290.77, s15.5, CH_3_0.77, s15.4, CH_3_0.89, s21.9, CH_3_300.87, s16.1, CH_3_0.87, s15.9, CH_3_1.07, s28.2, CH_3_313.43, m56.4, CH_2_171.2, C321.16, t (7.1)16.1, CH_3_2.07, s21.7, CH_3_Fig. 2^1^H^1^H COSY and key HMBC correlations of compounds 1–2, 8, 13–16, and 19–21
The NOESY correlations of H-3 with H-5, H-5 with H-9, H-17 with H_3_-30, and H_3_-18 with H_3_-19/H-13 suggest that** 1** is with a common dammarane-type triterpene skeleton. These correlations also support a β-orientation of the hydroxy group at C-3 (Fig. 3). Subsequently, the absolute configuration of 1 was determined by X-ray crystal diffraction as 3S, 5R, 8R, 9R, 10R, 13R, 14R, 17S, 24R (Fig. 4). Thus, the structure of compound 1 was assigned as shown in Fig. 1.Fig. 3. Key NOESY correlations of compounds 1–2, 8, 13–16, and 19–21Fig. 4ORTEP drawings of compounds 1, 2, 8, 13, and 14
Aglodorol B (2) was isolated as colorless needle crystals, mp 165–167 °C, with a molecular formula of C_32_H_56_O_3_ based on HRESIMS data (m/z 489.4301 [M + H]^+^, calcd for C_32_H_57_O_3_, 489.4308). The IR spectrum indicates the presence of hydroxy group (3413 cm^− 1^) and double bond (1638 cm^− 1^).
The ^1^H NMR data present seven methyl singlet [δH 1.14 (H_3_-27), 1.11 (H_3_-26), 0.98 (H_3_-28), 0.97 (H_3_-18), 0.87 (H_3_-30), 0.84 (H_3_-19), and 0.77 (H_3_-29)], a group of ethyloxyl [δH 1.16 (3H, t, J = 7.1 Hz, H_3_-32), 3.34 (2H, m, H_2_-31)], and two terminal olefinic methylene [δH 4.76 and 4.71 (s, H_2_-21)] protons (Table 1). The analysis of ^13^C NMR and HSQC spectra of compound 2 shows 32 carbon signals, including eight methyl, 12 methylene, six methine, and six quaternary carbons. The ^1^H and ^13^C NMR data of 2 are comparable to those of the reported compound 4 [9], except that the OCH_3_ at C-25 is replaced by the OCH_2_CH_3_ in 2, which is confirmed by the ^1^H–^1^H COSY correlations of H_2_-31/H_3_-32 and the HMBC correlations from H_2_-31 to C-25 (Fig. 2). Thus, the planar structure of 2 was established.
Similarly, the NOESY correlations of H-3 with H-5, H-5 with H-9, H-17 with H_3_-30, H_3_-18 with H_3_-19, and H-13 with H_3_-18, which are consistent with those of the dammarane-type triterpene skeleton, supported the β-orientation of the hydroxy group at C-3 (Fig. 3). Subsequently, the absolute structure of 2 was determined as 3S, 5R, 8R, 9R, 10R, 13R, 14R, 17S, 24S by a single crystal X-ray diffraction experiment (Fig. 4). Considering the use of ethanol in the extraction procedure and the possibility of transetherification of 4 to 2 during ethanol extraction, we performed a rapid extraction of plant material using methanol and analyzed it by UPLC-Qtrap-MS/MS (Fig. S22). However, there is no detectable signal for compound 2 in the extract. This result provides evidence that compound 2 may be naturally present in the plant at a level below the detectable level. However, this result could not exclude the possibility that compound** 2** may be formed from the methoxy-substituted analog of compound 4 via an acid-catalyzed transetherification during ethanol extraction.
Aglodorol C (8) was obtained as colorless needle crystals, mp 241–242 °C. Its molecular formula is C_32_H_50_O_7_ based on HRESIMS data (m/z 591.3533 [M + HCOO]^−^, calcd for C_33_H_51_O_9_, 591.3528), requiring eight degrees of unsaturation. The IR spectrum shows signals of hydroxy group (3456 cm^− 1^) and ester carbonyl group (1724, 1767 cm^− 1^).
The ^1^H NMR spectrum shows eight methyl singlets [δH 2.07 (H_3_-32), 1.34 (H_3_-26), 1.29 (H_3_-27), 1.07 (H_3_-30), 1.06 (H_3_-18), 0.90 (H_3_-19), 0.89 (H_3_-29), and 0.85 (H_3_-28)], one olefinic proton [δH 5.49 (1H, d, J = 2.9 Hz, H-15)], and four oxygenated methine protons [δH 4.65 (1H, m, H-3), 4.62 (1H, m, H-23), 3.92 (1H, t, J = 2.9 Hz, H-7), and 3.29 (1H, d, J = 1.3 Hz, H-24)] (Table 1). The ^13^C NMR spectrum of compound 8 shows 32 carbon signals, which are further classified by HSQC experiments as eight methyl, seven methylene, nine methine, and eight quaternary carbons. According to the chemical shifts, two olefinic carbons [δC 161.6 (C-14), 119.4 (C-15)], five oxygenated carbons [δC 78.2 (C-3), 77.6 (C-23), 76.1 (C-24), 72.7 (C-25), 72.4 (C-7)], and two ester carbonyl carbons [δC 171.2 (C-31), 177.9 (C-21)] could be determined. The NMR data of 8 are similar to those of agladoral A, an apotirucallane-type triterpene isolated from this genus, except that one group of the acetoxyl protons in agladoral A has disappeared and a carbonyl carbon (δC 177.9) in 8 substitutes a hydroxyl carbon (δC 97.1) in agladoral A [19]. The HMBC correlations from H-3 to C-31 and from H_3_-32 to C-31 indicate that the acetoxy group is located at C-3 (Fig. 2). The ^1^H–^1^H COSY correlations of H-17/H-20/H_2_-22/H-23/H-24, as well as the HMBC correlations from H-20 and H_2_-22 to C-21, from H_2_-22 to C-20/C-23, and from H-20 to C-17/C-13/C-16 confirm the presence of a five-membered lactone ring linked at C-17 (Fig. 2).
Similar to agladoral A, the NOESY correlations of H-3 with H_3_-28/H_3_-29, H-7 with H_3_-30, and H_3_-30/H_3_-19 with H-11β support the β-orientation of H-3 and H-7 (Fig. 3). Moreover, the NOESY correlations of H-17 with H-12a, H_3_-18 with H-20, and H-20 with H-23 illustrate the α-orientation of H-20 and H-23 in 8 (Fig. 3). The small coupling constant (J23, 24 = 1.27 Hz) indicates that they are in a gauche relationship [20], indicating that H-24 is β-oriented. Due to a suboptimal Flack parameter of 0.13(15) obtained from the single crystal X-ray diffraction experiment, only the relative configuration of compound 8 could be determined (Fig. 4 and Table S1). Consequently, the absolute configuration was subsequently determined as 3S, 5R, 7R, 8R, 9R, 10S, 13S, 17S, 20S, 23R, 24S by the ECD calculation (Fig. 8).
Aglodorol D (13), obtained as colorless massive crystals, mp 152–153 °C, has a molecular formula of C_20_H_33_O_2_ with four indices of hydrogen deficiency based on the HRESIMS (m/z 305.2480 [M − H]^−^, calcd for 305.2481) and supported by ^13^C NMR data. The IR spectrum shows the absorptions at 3326 cm^− 1^ for hydroxy group and 1705 cm^− 1^ for double bond.
The ^1^H NMR data present five methyl singlets [δH 1.68 (H_3_-15), 1.27 (H_3_-16), 1.19 (H_3_-18), 1.17 (H_3_-19), 0.83 (H_3_-20)] (Table 2). The analysis of ^13^C NMR and HSQC spectra of compound 13 reveals 20 carbon signals, including five methyl, six methylene, five methine, and four quaternary carbons. Among them, two olefinic (δC 134.2, 124.7) and two oxygenated (δC 76.8, 74.2) carbons are easily recognized. The alkene bond accounts for one of the four degrees of unsaturation, and the left three ones indicate that 13 contains a tricyclic system. The ^1^H–^1^H COSY correlations of H_2_-2/H_2_-1/H-14/H-5/H-4, and the HMBC correlations from H-4 to C-2/C-5/C-14/C-15, from H_2_-1 to C-3/C-5/C-14, from H-5 to C-3, and from H_3_-15 to C-2/C-3/C-4 suggest the presence of a six membered ring A and a methyl is linked to C-3 (Fig. 2). Another spin system, H_2_-7/H_2_-8/H-9/H-10, is elucidated by analysis of the ^1^H–^1^H COSY spectrum, and the HMBC correlations from H-10 to C-6/C-9, from H-7a to C-9/C-10, and from H_2_-8 to C-6/C-10 construct a five membered ring B (Fig. 2). Besides, the ^1^H–^1^H COSY correlations of H-10/H-11a/H-12a and the HMBC correlations from H-12b and H-14 to C-13 define the seven-membered ring C (Fig. 2). The HMBC correlations of H_2_-1 with C-13, H-14 with C-1/C-13, and H-5 with C-3/C-4/C-6/C-10 indicate that C-5 and C-14 are shared by the six- and seven-membered rings. While, the HMBC correlations of H-9 with C-11, H-10 with C-5, H-7 with C-5/C-6, and H_3_-20 with C-5/C-6/C-7 indicate that C-6 and C-10 are shared by five- and seven-membered rings (Fig. 2). Hereby, 13 was deduced to have a 6/7/5 membered ring system. The presence of the 2-hydroxypropan-2-yl is indicated by the HMBC correlations of H_3_-18 and H_3_-19 with C-9/C-17 (δC 74.2), and its position is fixed on C-9 of the five-membered ring. The HMBC correlations of H_3_-16 [δH 1.27 (3H, s)] with C-12, C-13, and C-14, and of H_3_-20 with C-5, C-6, C-7, and C-10 deduce the positions of two aliphatic methyl groups at C-13 and C-6, respectively. At the same time, in combination of the chemical shift of C-13 (δC 76.8) and the formula weight, a hydroxy is determined to be located at C-13. The planar structure of 13 was thus established. Table 2^1^H (500 MHz) and ^13^C (125 MHz) NMR data of compounds 13, 14, 15, 16, 19, 20, and 21 (δ in ppm, J in Hz) in Chloroform-d_1_No.13141516192021δH (J in Hz)δC, typeδH (J in Hz)δC, typeδH (J in Hz)δC, typeδH (J in Hz)δC, typeδH (J in Hz)δC, typeδH (J in Hz)δC, typeδH (J in Hz)δC, type11.53, m22.4, CH_2_150.3, C43.6, C44.0, C46.8, C45.9, C44.4, C2α1.98, m31.4, CH_2_2.08, m35.8, CH_2_1.39, m39.4, CH_2_1.39, m35.7, CH_2_2.09, m42.5, CH_2_1.98, dd (13.6, 11.9)42.4, CH_2_1.72, m49.5, CH_2_2β2.29, m1.79, m1.75, dd (14.5, 2.9)1.68, dd (13.6, 3.9)3α134.2, C1.84, m32.3, CH_2_1.47, m51.7, CH1.76, m49.5, CH5.31, dd (11.5, 2.9)123.9, CH5.07, dd (11.9, 3.9)124.7, CH4.37, t (6.0)72.8, CH3β1.47, m45.39, d (5.3)124.7, CH3.32, dd (11.8, 4.4)77.9, CH80.7, C82.9, C135.0, C132.6, C151.9, C52.23, m47.4, CH44.7, C1.80, m38.9, CH_2_1.65–1.67, m38.5, CH_2_2.15, m37.4, CH_2_2.23–2.25 m39.7, CH_2_2.25, m/2.06, m25.2, CH_2_6α50.9, C1.57, m35.3, CH_2_1.86, m24.7, CH_2_1.68, m28.8, CH_2_2.21, m34.1, CH_2_3.40, m34.0, CH_2_2.04, m32.8, CH_2_6β2.12, m1.64, m1.74, m2.11, m1.96, m1.93, m7α1.56, m42.3, CH_2_2.02, m29.0, CH_2_2.04, m51.7, CH3.32, q (8.9)49.3, CH3.87, dd (8.2, 2.5)75.2, CH206.3, C4.24, dd (8.3, 3.4)71.8, CH7β1.35, m81.37, m25.5, CH_2_1.65, m47.9, CH152.5, C152.8, C153.4, C150.1, C150.9, C9α1.63, m56.8, CH1.78, m57.9, CH1.96, m32.8, CH_2_2.26, m31.6, CH_2_2.01, m34.7, CH_2_2.49 (m),28.6, CH_2_2.26, m34.0, CH_2_9β2.34, m2.02, m1.90, m2.35 (m)2.09, m10a2.24, m47.5, CH1.86, m26.0, CH_2_1.53, m32.1, CH_2_1.48, m32.4, CH_2_1.50, m29.2, CH_2_1.21 (m)31.3, CH_2_1.29, m30.2, CH_2_10b1.44, m1.97, m1.89, m1.84, m1.39 (m)1.71, m11α1.19, m28.2, CH_2_1.34, m44.2, CH_2_1.64, m41.5, CH1.64, m42.0, CH1.42, m43.8, CH1.28 (m)42.2, CH1.59, m47.9, CH11β2.11, m12α1.82, m31.7, CH_2_45.7, C1.75, m53.4, CH1.74, m53.4, CH1.67, m59.4, CH1.52 (m)59.5, CH2.45, m57.2, CH12β1.59, m13a76.8, C1.74, m43.6, CH_2_1.74, m25.1, CH_2_1.75, m25.2, CH_2_1.28, m27.1, CH_2_1.26 (m)27.4, CH_2_1.38, m28.8, CH_2_13b1.49, m1.34, m1.38, m1.69, m1.62 (m)1.86, m14α1.66, m51.8, CH1.93, m46.9, CH1.16, m40.0, CH_2_1.10, m38.5, CH_2_1.42, m44.2, CH_2_1.29 (m)43.7, CH_2_1.41, m44.3, CH_2_14β1.42, m1.51, m1.49, m15a1.68, s23.9, CH_3_4.79, s107.9, CH_2_0.93, s23.2, CH_3_0.87, s23.2, CH_3_1.02, s21.8, CH_3_0.83 (s)21.9, CH_3_0.93, s18.4, CH_3_15b4.61, s161.27, s34.2, CH_3_1.00, s18.6, CH_3_1.13, s24.4, CH_3_1.26, s24.9, CH_3_1.59, s16.4, CH_3_1.60(s)15.9, CH_3_5.14, s/4.94, s110.7, CH_2_17a74.2, C73.9, C4.95, br. s109.1, CH_2_4.94, s113.4, CH_2_5.18, s108.3, CH_2_6.01 (s),126.0, CH_2_5.22, s113.6, CH_2_17b4.84, br. s4.82, s5.13, s5.88 (s)4.96, s181.19, s29.3, CH_3_1.18, s29.1, CH_3_74.6, C74.4, C74.0, C71.6, C150.2, C19a1.17, s26.8, CH_3_1.18, s27.2, CH_3_1.14, s27.2, CH_3_1.16, s27.7, CH_3_1.19, s25.2, CH_3_1.00 (s)25.7, CH_3_4.78, s110.6, CH_2_19b4.66, s200.83, s16.9, CH_3_0.75, s9.5, CH_3_1.15, s29.1, CH_3_1.17, s28.9, CH_3_1.25, s31.6, CH_3_1.12 (s)31.8, CH_3_1.74, s19.3, CH_3_18–OH4.15 (s)
The relative configuration of 13 was determined by the NOESY spectrum (Fig. 3). The NOESY correlations of H-14 with H-5/H-10 suggest that these protons are on the same side of the molecule, while the correlations of H_3_-20 with H-9/H_3_-16 suggest that they are on the other side of the molecule. Therefore, a cis-fusion between the six- and seven-membered rings and a trans-fusion between the seven- and five-membered rings were deduced. Subsequent single crystal X-ray diffraction analysis assigned the absolute configuration of 13 as 5R, 6R, 9S, 10R, 13S, 14S (Fig. 4).
Aglodorol E (14) was obtained as colorless needle crystals, mp 145–146 °C. Its molecular formula was deduced as C_20_H_34_O_2_ based on the HRESIMS (m/z 305.2480, [M − H]^−^, calcd for 305.2481) and ^13^C NMR data with the presence of four hydrogen-deficiency indices. The IR absorption bands at 3385 cm^− 1^ and 1641 cm^− 1^ reveal the presence of hydroxy group and double bond.
The ^1^H NMR data include four methyl singlets [δH 1.18 (H_3_-18 and H_3_-19), 1.00 (H_3_-16), 0.75 (H_3_-20)], and a pair of terminal olefinic methylene signals [δH 4.79 (1H, s), 4.61 (1H, s)] (Table 2). The analysis of ^13^C NMR data and HSQC spectrum of compound 14 reveals 20 carbon signals, including four methyl, eight methylene, four methine, and four quaternary carbons. Among them, two olefinic carbons (δC 150.3, 107.9) and two oxygenated carbons (δC 77.9, 73.9) are particularly obvious. Similar to 13, compound 14 should also contain a tricyclic system, since the alkene bond accounts for only one of the four degrees of unsaturation. The ^1^H–^1^H COSY correlations of H_2_-2/H_2_-3/H-4 and the HMBC correlations from H_2_-15 to C-14 and C-2, from H-4 to C-3 and C-5, and from H-14 to C-5 correspond to a six membered ring and an exomethylene group is defined at C-1 and C-15 (Fig. 2). The COSY cross peaks of H-8/H-9//H_2_-10/H_2_-11, and the HMBC correlations from H_2_-11 to C-9/C-10/C-12, from H-8 to C-11/C-12, and from H_3_-16 to C-8/C-11/C-12 define the presence of a five membered ring. In addition, the COSY correlations of H-13a/H-14 and H-6a/H_2_-7/H-8, along with the HMBC correlations of H-8 and H-14 with C-13, and H-6 with C-5/C-14 confirm the existence of a seven membered ring (Fig. 2). The connections of the six membered ring with the seven membered ring, and the seven membered ring with the five membered ring are established by the HMBC correlations of H_3_-20 with C-4/C-5/C-6/C-14, H_3_-16 with C-8/C-11/C-12/C-13, H-4 with C-6, H-9 with C-7, and H-11 with C-13. This led to the determination of the planar structure of 14.
The relative configuration of 14 was established through NOESY analysis: key correlations of H-14 with H-4/H_3_-16 confirmed that they are on the same side of the fused ring system, tentatively on the β-orientation, whereas the observed NOESY interaction of H-8 with H_3_-20 positions them on the opposing α-orientation (Fig. 3). The absolute configuration of 14 was further determined by X-ray crystal diffraction to be 4R, 5R, 8R, 9S, 12S, 14S (Fig. 4).
Aglodorol F (15) was obtained as colorless solid. Its molecular formula was determined to be C_20_H_34_O_2_ by HRESIMS (m/z 351.2539 [M + HCOO]^−^, calcd. for C_21_H_35_O_4_, 351.2530). The IR absorption bands at 3405 cm^− 1^ and 1635 cm^− 1^ indicate the presence of hydroxy group and double bond.
The ^1^H NMR data of 15 reveal two distinct terminal olefinic protons [δH 4.95 (1H, br. s), 4.84 (1H, br. s), H-17] and four methyl groups [δH 1.15 (H_3_-20), 1.14 (H_3_-19), 1.13 (H_3_-16), 0.93 (H_3_-15)] (Table 2). The ^13^C NMR and HSQC spectra resolve 20 carbon resonances attributable to two olefinic [δC 152.5 (C-8), 109.1 (C-17)], four methyls [δC 29.1 (C-20), 27.2 (C-19), 24.4 (C-16), 23.2 (C-15)], seven methylene [δC 40.0 (C-14), 39.4 (C-2), 38.9 (C-5), 32.8 (C-9), 32.1 (C-10), 25.1 (C-13), 24.7 (C-6)], four common methine [δC 53.4 (C-12), 51.7 (C-3 and C-7), 41.5 (C-11)], two oxygenated quaternary [δC 80.7 (C-4), 74.6 (C-18)], and one quaternary [δC 43.6 (C-1)] carbons. The olefinic moiety accounts for one of four degrees of unsaturation, so there may be a tricyclic skeleton in the structure of 15, which is further confirmed by the HMBC and ^1^H–^1^H COSY correlations (Fig. 2). The ^1^H–^1^H COSY correlations of H_2_-5/H_2_-6, H-3/H-7 and the HMBC correlations from H-3 to C-4/C-16, from H_3_-16 to C-3/C-5, from H-5 to C-3/C-4/C-7/C-16, from H-6 to C-4/C-5/C-7, and from H-7 to C-3 correspond to a five membered ring with a methyl group at C-4. The ^1^H–^1^H COSY correlations of H_2_-9/H_2_-10/H-11 and the HMBC correlations from H_2_-2 to C-1/C-3/C-11, from H-7 to C-3/C-8/C-9, from H_2_-17 to C-7/C-8/C-9, from H_2_-9 to C-7/C-8/C-10/C-11/C-17, from H_2_-10 to C-1/C-9/C-11, and from H-11 to C-10 define the presence of an eight membered ring and confirm that the terminal double bond is at the C-8 and C-17. In addition, the ^1^H–^1^H COSY correlations (H-11/H-12/H_2_-13/H_2_-14) and the HMBC correlations (from H-12 to C-1/C-11/C-14, from H_2_-13 to C-1/C-11/C-14, and from H_2_-14 to C-11) confirm the existence of another five membered ring. The 2-hydroxypropan-2-yl is at C-12 as evidenced by the HMBC correlations from H_3_-20 and H_3_-19 to C-12/C-18. The above tricyclic skeleton is subsequently established through the following key HMBC correlations: H-3 to C-2/C-4/C-5, H_3_-15 to C-1/C-2/C-11/C-14, and H_2_-2 to C-14. Thus, the planar structure of compound 15 was determined. It is interesting to note that 15 has the same planar structure as compound 17 [21], but their NMR spectra exhibited significant discrepancies. Notable chemical shift variations (Δδ = 2–6 ppm) for the eight membered ring carbons and C-12/15 may originate from alterations in the chiral configurations at specific positions (C-1, C-3, C-7, C-11, and C-12).
Consistent with compound 17, the NOESY correlations (Fig. 3) of H-12/H_3_-15 and H-11/H_3_-19 identified the 1R**, 11S**, 12R**-configurations [21]. The NOESY correlation between H_3_-16 and H-7 indicates their cofacial orientation, and on the basis of above, their relative configurations may be as 4R**, 7S** or 4S**, 7R**. However, the configuration at C-3 could not be determined due to the absence of valuable NOESY correlations observed. To establish the relative configuration of compound 15, the quantum chemical calculation of NMR parameters of four epimers 15a (1R**, 3S**, 4R**, 7S**, 11S**, 12R**), 15b (1R**, 3R**, 4R**, 7S**, 11S**, 12R**), 15c (1R**, 3R**, 4S**, 7R**, 11S**, 12R**), and 15d (1R**, 3S**, 4S**, 7R**, 11S**, 12R**) were performed, and the result showed that the configuration of 15b is more consistent with the experimental value of 15 (Fig. 5). Furthermore, the absolute configuration of 1S*, 3S, 4S, 7R, 11R, 12S was suggested by the ECD calculation (Fig. 8).Fig. 5^13^C-NMR calculations of compound 15
Aglodorol G (16) was obtained as colorless solid. Its molecular formula was determined as C_20_H_34_O_2_ based on HRESIMS data (m/z 351.2536 [M + HCOO]^−^, calcd. for C_21_H_35_O_4_, 351.2541), which is the same as that of compound 15. The IR absorption band at 3389 cm^− 1^ indicates the presence of hydroxy group.
The ^1^H NMR data of 16 reveal two distinct terminal olefinic protons [δH 4.94 (1H, br. s), 4.82 (1H, br. s)] and four methyl groups [δH 1.26 (H_3_-16), 1.17 (H_3_-20), 1.16 (H_3_-19), 0.87 (H_3_-15)] (Table 2). The ^13^C NMR and HSQC spectra resolve 20 carbon resonances attributable to two olefinic [δC 152.8 (C-8), 113.4 (C-17)], four methyl [δC 28.9 (C-20), 27.7 (C-19), 24.9 (C-16), 23.2 (C-15)], seven methylene [δC 38.5 (C-14 and C-5), 35.7 (C-2), 32.4 (C-10), 31.6 (C-9), 28.8 (C-6), 25.2 (C-13)], four methine [δC 53.4 (C-12), 49.5 (C-3), 49.3 (C-7), 42.0 (C-11)], two oxygenated quaternary [δC 82.9 (C-4), 74.4 (C-18)], and one quaternary [δC 44.0 (C-1)] carbons. Compound 16 was deduced to be an isomer of compound 15 because of their very similar 1D NMR, HMBC and COSY data (Table 2 and Fig. 2).
The NOESY correlations of H_3_-15 with H-12/H_3_-16/H-9a indicate that they are in the same direction in space, tentatively assigned as the β-orientation, while the NOESY correlations of H-11 with H_3_-19 and H-7 with H-9b suggest that they are in the other direction of the ring system (Fig. 3). Since the configuration at C-3 could not be deduced from the NOESY correlations, the quantum chemical calculations of two epimers 16a (1R**, 3R**, 4R**, 7R**, 11S**, 12R**) and 16b (1R**, 3S**, 4R**, 7R**, 11S**, 12R**) were performed. The results showed that the configuration of 16a is more consistent with the experimental data (Fig. 6). Furthermore, the absolute configuration of 1S, 3S, 4S, 7S, 11R, 12S was suggested by the ECD calculation (Fig. 8).Fig. 6^13^C-NMR calculations of compound 16
Aglodorol H (19) was obtained as colorless oil with a molecular formula of C_20_H_34_O_2_ as evidenced by HRESIMS (m/z 305.2487 [M − H]^−^, calcd. for C_20_H_33_O_2_, 305.2486). The IR absorption bands at 3425 cm^− 1^ and 1663 cm^− 1^ reveal the presence of hydroxy group and double bond.
The ^1^H NMR data of 19 reveal three olefinic protons [δH 5.18 (1H, br.s), 5.13 (1H, br. s), 5.31 (1H, dd, J = 11.5, 2.9 Hz)], four methyl groups [δH 1.59 (H_3_-16), 1.25 (H_3_-20), 1.19 (H_3_-19), 1.02 (H_3_-15)], and one oxygenated methine proton [δH 3.87 (1H, dd, J = 8.2, 2.5 Hz, H-7)] (Table 2). The ^13^C NMR and HSQC spectra resolve 20 carbon resonances attributable to four olefinic [δC 153.4 (C-8), 135.0 (C-4), 123.9 (C-3), 108.3 (C-17)], four methyl [δC 31.6 (C-20), 25.2 (C-19), 21.8 (C-15), 16.4 (C-16)], seven common methylene [δC 44.2 (C-14), 42.5 (C-2), 37.4 (C-5), 34.7 (C-9), 34.1 (C-6), 29.2 (C-10), 27.1 (C-13)], one oxygenated methine [δC 75.2 (C-7)], two methine [δC 59.4 (C-12), 43.8 (C-11)], one oxygenated quaternary [δC 74.0 (C-18)], and one quaternary [δC 46.8 (C-1)] carbons. The two olefinic bonds account for two of the four degrees of unsaturation, so there may be a bicyclic backbone in the structure of 19. The ^1^H–^1^H COSY correlations (H_2_-2/H-3, H_2_-5/H_2_-6/H-7 and H_2_-9/H_2_-10/H-11) and the HMBC correlations (from H_2_-2 to C-1/C-3/C-4, from H-3 to C-2, from H_2_-5 to C-3/C-4/C-16, from H_2_-6 to C-7/C-8/C-16/C-17, from H-7 to C-6/C-9/C-17, from H_2_-9 to C-8, and from H_2_-10 to C-1) correspond to an eleven membered ring with a methyl group at C-4 and a terminal double bond at C-8 (Fig. 2). In addition, the ^1^H–^1^H COSY correlations (H-12/H_2_-13/H_2_-14) and the HMBC correlations (from H-11 to C-1/C-12/C-13) confirm the existence of a five membered ring. The 2-hydroxypropan-2-yl is at C-12 as evidenced by the HMBC correlations from H_3_-20 and H_3_-19 to C-12/C-18 and from H-11 to C-18. The above bicyclic skeleton is deduced to be connected via C-1 and C-11 as evidenced by the key HMBC correlations of H_2_-2 to C-14 and H_3_-15 to C-1/C-2 (Fig. 2).
The NOESY correlations of H_3_-15 with H-12 suggest that they are on the same side of the molecule, while the correlations of H-11 with H_3_-18 suggest that they are on the other side of the molecule (Fig. 3). The E-configuration of the Δ^3^ double bond is deduced by the NOESY correlations of H-3 with H_2_-5 [16]. In order to establish the relative configuration of 7-OH, the quantum chemical calculations were carried out on 19 (Fig. 7), and the (1R**, 3E*, 7S**, 11S**, 12R**)-configuration (19b) was determined. Furthermore, the ECD calculation suggests the absolute configuration of 19 as 1S*, 3E, 7R, 11R, 12S (Fig. 8).Fig. 7^13^C-NMR calculations of compounds 19 and 21Fig. 8. The experimental and calculated ECD spectra of compounds 8, 15–16, and 19–21
Aglodorol I (20) was obtained as light-yellow oil with a molecular formula of C_20_H_32_O_2_ and five degrees of unsaturation based on HRESIMS (m/z 303.2327 [M − H]^−^, calcd. for C_20_H_31_O_2_, 303.2324). The 1D and 2D NMR spectra suggest that the structure of 20 is very similar to that of compound 19. The only difference is that the hydroxy group at C-7 (δC 75.2) in 19 is oxidized to a carbonyl group (δC 206.3) in 20 (Table 2). The absolute configuration of 20 was determined as 1S, 3E, 11R, 12S by the ECD calculation (Fig. 8).
Aglodorol J (21) was obtained as colorless oil with a molecular formula of C_20_H_32_O_2_ and five degrees of unsaturation based on the HRESIMS (m/z 303.2325 [M − H]^−^, calcd. for C_20_H_31_O_2_, 303.2330). The IR spectrum shows stretching bands of hydroxy group (3396 cm^− 1^) and double bond (1640 cm^− 1^).
The ^1^H NMR data of 21 present three pairs of terminal double bond protons [δH 5.22 (1H, s, H-17), 5.14 (1H, s, H-16), 4.96 (1H, s, H-17), 4.94 (1H, s, H-16), 4.78 (1H, s, H-19), 4.66 (1H, s, H-19)], two methyl singlets [δH 1.74 (H_3_-20), 0.93 (H_3_-15)], and two oxygenated methine protons [δH 4.37 (1H, t, J = 6.0 Hz, H-3), 4.24 (1H, dd, J = 8.3, 3.4 Hz, H-7)] (Table 2). The ^13^C NMR and HSQC spectra resolve 20 carbon resonances attributable to six olefinic [δC 151.9 (C-4), 150.9 (C-8), 150.2 (C-18), 113.6 (C-17), 110.7 (C-16), 110.6 (C-19)], two methyl [δC 19.3 (C-20), 18.4 (C-15)], seven methylene [δC 49.5 (C-2), 44.3 (C-14), 34.0 (C-9), 32.8 (C-6), 30.2 (C-10), 28.8 (C-13), 25.2 (C-5)], two oxygenated methine [δC 72.8 (C-3), 71.8 (C-7)], two methine [δC 57.2 (C-12), 47.9 (C-11)], and one quaternary [δC 44.4 (C-1)] carbons. The olefinic moieties account for three degrees of unsaturation, so there should be a bicyclic backbone in the structure of 21. The ^1^H–^1^H COSY correlations (H_2_-2/H-3, H_2_-5/H_2_-6/H-7, H_2_-9/H_2_-10/H-11/H-12/H_2_-13/H_2_-14) and the HMBC correlations of H-3 with C-2/C-4/C-5/C-16, H_2_-16 with C-3/C-5, H_2_-17 with C-7/C-8/C-9, H_2_-9 with C-8/C-7/C-11, H_2_-10 with C-11/C-1, H_3_-15 with C-1/C-2/C-11/C-14 and H_2_-14 with C-11/C-12/C-15 indicate the bicyclic skeleton of 21 as shown in Fig. 2. An isopropenyl group is located at C-12, confirmed by the HMBC correlations of CH_3_-20/H_2_-19 with C-12. The planar structure of 21 is consistent with that of dolabella-4(16), 8(17), 18(19)-triene-3,7-diol [5]. However, the differences of their NMR data indicate the differences in their stereo structures.
The relative configuration of 21 was determined as 1R**, 3S**, 7S**, 11S**, 12R** (21a) by the NOESY correlations (H_3_-15 with H-12, H-3 with H-7, and H-11 with H_3_-20) (Fig. 3) and the quantum chemical calculations (Fig. 7). Further, the absolute configuration of 1S*, 3R, 7R, 11R, 12S was determined by comparison of the experimental ECD and calculated ECD curves (Fig. 8).
Compounds 17, 18, and 22 have only been reported with relative configurations previously; here, we determined their absolute configurations by comparison of calculated and experimental ECD results (Fig. S98).
Investigation neuroprotective activity
The crude extract and several secondary metabolites of A. odorata have successively been reported to demonstrate neuroprotective properties in models of cerebral ischaemia–reperfusion injury and neuroinflammation [4, 5]. Thus, we evaluated the isolated compounds in four neurodegeneration cell models.
The results showed that compounds 3,** 9**–11, 13, and 17–18 exhibited significant neuroprotective effects against OGD/R-mediated nerve injury in PC12 cells at 10 µM concentration (P < 0.05) (Fig. 9). Compounds 3, 8, and 19 showed significant protective effects against L-glutamate-induced excitatory damage at a concentration of 20 µM (Fig. 10). In both models, the ortho-dihydroxy moiety may serve as a key pharmacophore for triterpenoid-mediated neuroprotection. Notably, at non-cytotoxic concentrations, compound 3 conferred protection against OGD/R injury, whereas structural analogs 1 and 5 exacerbated OGD/R-induced damage in PC12 cells. This observation suggests that chloro-substitution modification at C24 or reductive modification at C25 may potentiate damage mechanisms underlying OGD/R.Fig. 9. The protective activity of the isolates at 10 µM concentration on OGD/R-mediated nerve injury in PC12 cells. A The cell viability of PC12 cells by OGD/R-mediated nerve injury with the treatment of the isolates; B Toxicity test of compounds on PC12 cells. Eda (16 μM) was used as positive control; ##p < 0.01 compared with control group; *p < 0.05; **p < 0.01 compared with OGD/R group (n = 6)Fig. 10. The anti-excitotoxicity activity of the isolates in the L-glutamate-induced HT22 cells at a concentration of 20 µM. ###p < 0.001 compared with control group; *p < 0.05, **p < 0.01 compared with Glu group (n = 3)
In the LPS-induced neuroinflammation model, all tested compounds significantly suppressed NO production at a concentration of 20 µM (Fig. 11), with compound 22 demonstrating the highest potency (IC_50_ = 22.41 ± 0.32 µM) (Table 3). Compounds 1–2, 5–6, 8, 11, and 17 possessed remarkably significant anti-ferroptosis activity at 10 µM concentration (P < 0.001, Fig. 12), with the EC_50_ values of compounds 1, 2, 11, and 17 ranging from 1.16 to 9.45 µM (Table 3). In ferroptosis models, the terminal ortho-dihydroxy moiety is dispensable for triterpenoid activity. Strikingly, compounds 10 (inactive) and 11 (highly active), differing only at C-3, establish that the oxidation state at C-3 is critical for the biological activity of cycloartane-type triterpenoids.Fig. 11. The inhibitory activity of the isolates at a concentration of 20 μM on LPS-induced NO production in BV-2 cells. A The concentration of NO produced by LPS-induced BV2 cells with the treatment of the isolates; B Cell viability of BV-2 cells with the treatment of the isolates. Dex (10 μM) was used as positive control; ###p < 0.001 compared with control group; p < 0.01, *p < 0.001 compared with LPS group (n = 3)Table 3. Various activity data of active compoundsCom.IC_50_ (μM) ^a^EC_50_ (µM) ^a^NO inhibitionAnti-ferroptosis1NT^b^1.74 ± 0.112NT^b^1.36 ± 0.105 > 50NT^b^8 > 50NT^b^9 > 50NT^b^11NT^b^1.16 ± 0.0217NT^b^9.45 ± 0.392222.41 ± 0.32NT^b^Dex^c^6.78 ± 0.13–Fer-1^d^–0.0059 ± 0.00038^a^ IC_50_ and EC_50_ values are presented as mean ± SD (n = 3)^b^ NT, not tested^c^ Dex, Dexamethasone, positive control for LPS-induced NO production in BV2 cells^d^ Fer-1, Ferrostatin-1, positive control for RSL3-induced terroptotic in PC12 cellsFig. 12The anti-ferroptosis activity of the isolates at 10 µM concentration in RSL3-induced PC12 cell death. Fer-1 (10 μM) was used as positive control; ###p < 0.001 compared with control group; ***p < 0.001 compared with RSL3 group (n = 3)
Conclusion
In summary, the phytochemical investigation of A. odorata led to the successful isolation and identification of 22 terpenoids, including three new triterpenoids, seven novel diterpenoids, alongside 12 known compounds. Notably, diterpenoids are very rare in the genus Aglaia, particularly dolastane- and fusicoccane-type derivatives, which were previously documented predominantly in marine organisms. In four mechanistically distinct models of neuronal cell injury, triterpenoids showed superior neuroprotective activity compared to diterpenoids. This study not only expands the chemical diversity of terpenoids in the genus Aglaia, but also provides a mechanistic explanation for the neuroprotective activity of A. odorata against ischemic stroke through a multi-model evaluation system. These findings further provide a scientific basis for exploring this plant in anti-ischemic stroke drug discovery.
Experimental section
General experimental materials
UV spectra were recorded on a Shimadzu UV-2450 spectrophotometer (Shimadzu Co., Tokyo, Japan). Nicolet NEXUS-470 FTIR spectrometer (MA, USA) was used to record IR spectra. The NMR spectra were performed on an INOVA-500 NMR spectrometer (Varian Co., Palo Alto, CA, USA), using methanol-d4 and chloroform-d1 as solvents, and the chemical shifts were referenced to the solvent residual peak. HRESIMS data were performed on a Waters Xevo G2 Q-TOF MS (Waters Co., Milford, MA, UK) or an Orbitrap Exploris 240 (ThermoFisher). Optical rotations were measured using a Rudolph Autopol III automatic polarimeter (Rudolph Research, Fairfield, New jersey, USA). The experimental ECD spectra were obtained on a J-810 spectrophotometer (JASCO, Japan). HPLC was carried out on an Agilent 1260 series instrument with a Thermo ScientificTM AccucoreTM C18 5 μm column (250 × 9.4 mm). Single crystal diffraction data were measured on a XtaLAB Synergy R, HyPix diffractometer (Rigaku, Japan). Silica gel (100–200 mesh and 200–300 mesh, Qingdao Marine Chemical, Co., Ltd., P.R. China) and Sephadex LH-20 (18–110 μm, Pharmacia, Sweden) and ODS (50 μM, YMC, Japan) were used for open column chromatography (CC). TLC analyses were carried out on the precoated silica gel GF 254 plates (Qingdao Marine Chemical Co., Ltd., P.R. China).
Mouse BV-2 microglial cells, PC12 cells, and HT22 cells were sourced from Peking Union Medical College (Beijing, China). BV-2 cells and HT22 cells were maintained in high glucose Dulbecco’s Modified Eagle Medium (Gibico, GrandIsland, USA), while PC12 cells were maintained in RPMI 1640 medium (Gibico, GrandIsland, USA), both supplemented with 10% fetal bovine serum (ABW, Shanghai, China), penicillin and streptomycin (100 U/mL and 100U/mL). All cell lines were kept at 37 °C in a 5% CO_2_ in air humidified environment.
Methylthiazolyldiphenyl-tetrazolium bromide (MTT) was from Solarbio (Beijing, China). LPS was purchased from Sigma (St. Louis, MO, USA). Ferrostatin-1 (Fer-1) was supplied by MedChem Express (Monmouth Junction, NJ, USA). L-glutamate was offered by Shanghai Yuanye Biotechnology Co., Ltd. (Shanghai, China).
Plant material
The tender branches and leaves of Aglaia odorata Lour. were collected in Zhaoqing, Guangdong Province, China, in August 2020. And they were identified by one of the authors, Prof. PengFei Tu. The voucher specimen (No. 20200820) was deposited in Herbarium of the Modern Research Center for Traditional Chinese Medicine, Peking University.
Extraction and isolation
The air-dried and milled tender branches and leaves of A. odorata (6.8 kg) were extracted successively with 95% and 50% ethanol (3 × 60.0 L) under reflux condition. The filtrates were combined and evaporated under reduced pressure to obtain a crude extract (1.5 kg), which was suspended in water, and extracted sequentially with petroleum ether (PE), ethyl acetate (EA), and n-butanol. After concentration, the PE extract (260.0 g), EA extract (200.0 g), and n-butanol extract (250.0 g) were obtained.
The partial PE extract (80.0 g) was fractionated by silica gel CC (PE–acetone, 20:1 to 0:1, v/v) to yield ten fractions (PE1–PE10). PE4 (2.8 g) was separated by ODS CC (MeOH − H_2_O, 30:70 to 100:0, v/v) to obtain four fractions (PE4A–PE4F). PE4F (807.0 mg) was subjected to silica gel CC with a gradient elution of PE/acetone (20:1 to 0:1, v/v), to afford three fractions (PE4F1–PE4F3). PE4F3 (428.0 mg) was further purified by semi-preparative HPLC (MeCN–H_2_O, 75:25,* v*/v, 3 mL/min) to afford 1 (48.0 mg, tR 20.0 min) and 5 (50.0 mg, tR 26.1 min). PE5 (989.0 mg) was divided into four fractions (PE5A–PE5D) by a Sephadex LH-20 CC (CH_2_Cl_2_–MeOH, 1:1, v/v). PE5B (195.0 mg) and PE5C (358.0 mg) were chromatographed on an ODS column with a gradient of MeOH/H_2_O (from 50:50 to 100:0, v/v) to obtain five subfractions, PE5B1–PE5B5 and PE5C1–PE5C5, respectively. PE5B5 (27.0 mg) was further purified by semi-preparative HPLC (MeCN–H_2_O, 50:50, v/v, 3 mL/min) to yield 18 (3.0 mg, tR 25.0 min), 17 (2.0 mg, tR 23.0 min), and 22 (2.0 mg, tR 39.1 min). PE5C5 (102.0 mg) was then purified by semi-preparative HPLC (MeCN–H_2_O, 85:15,* v*/v, 3 mL/min) to yield 11 (18.0 mg, tR 16.0 min). PE6 (5.4 g) was similarly separated on a Sephadex LH-20 (CH_2_Cl_2_–MeOH, 1:1, v/v) to yield three fractions (PE6A–6C). PE6B (3.0 g) was chromatographed on an ODS column (MeOH − H_2_O, 3:7 to 10:0, v/v) to obtain six subfractions (PE6B1−PE6B6). PE6B5 (2.3 g) yielded a large amount of precipitate (0.71 g), which was purified via semi-preparative HPLC (MeCN–H_2_O, 85:15, 3 mL/min) to obtain compounds 3 (105.0 mg, tR 25.0 min) and 6 (10.0 mg, tR 29.0 min). PE6B4 (630.0 mg) and the supernatant of PE6B5 (1.6 g) were further separated by ODS CC (MeOH−H_2_O, 50:50 to 100:0, v/v), both obtaining five sub fractions, PE6B4A−PE6B4E and PE6B5A−PE6B5E. Compound 13 (5.0 mg, tR 38.0 min) was obtained from PE6B4E (50.0 mg) by semi-preparative HPLC (MeCN–H_2_O, 52:48, 3 mL/min). Compound 14 (2.0 mg, tR 41.3 min) was obtained from PE6B4D (261.0 mg) by semi-preparative HPLC (MeCN–H_2_O, 45:55, v/v, 3 mL/min). Compound 9 (2.0 mg, tR 22.0 min) and 10 (23.0 mg, tR 28.0 min) were purified from PE6B5B (69.0 mg) by semi-preparative HPLC (MeCN–H_2_O, 65:35, v/v, 3 mL/min).
The EA extract (200.0 g) was fractionated by silica gel CC (PE–EA, 99:1 to 0:1, v/v) to yield ten fractions (EA1–EA10). EA3 (18.0 g), EA4 (32.0 g), and EA5 (48.0 g) were separated by MCI-GEL CHP20P CC and eluted with a stepwise gradient of MeOH/H_2_O (5:95, 30:70, 50:50, 70:30, 90:10 and 100:0, v/v) to yield four fractions (EA3A–EA3D), five fractions (EA4A–EA4E), and seven fractions (EA5A–EA5G), respectively. EA3A was subjected to a silica gel column eluted with PE/CH_2_Cl_2_ (1:5, 1:7, 1:10 and 1:20, v/v) to obtain four fractions (EA3A1–EA3A4). EA3A4 was further fractionated by ODS CC (MeOH–H_2_O, 30:70 to 100:0, v/v) to yield four fractions (EA3A4A–EA3A4D). EA3A4D was finally purified by semi-preparative HPLC (MeCN–H_2_O, 55:45, v/v, 3 mL/min) to yield 20 (12.0 mg, tR 23.5 min). EA3C was purified by semi-preparative HPLC (MeCN–H_2_O, 78:22,* v*/v, 3 mL/min) to afford** 4** (6.0 mg, tR 30.7 min) and 7 (9.0 mg, tR 36.1 min). EA4D was chromatographed on ODS column (MeOH–H_2_O, 30:70, 50:50, 70:30, 100:0, v/v) to obtain 14 fractions (EA4D1–EA4D14). EA4D14 was purified by semi-preparative HPLC (MeCN–H_2_O, 88:12, v/v, 3 mL/min) to yield 2 (13.0 mg, tR 23.7 min). EA5E was processed by a Sephadex LH-20 column (CH_2_Cl_2_–MeOH, 1:1, v/v) to obtain nine subfractions (EA5E1–EA5E9). Compound 12 (3.0 mg, tR 25.3 min) was obtained from EA5E7 by semi-preparative HPLC (MeCN/H_2_O, 90:10,* v*/v, 3 mL/min). EA6 (800.0 mg) and PE7 (750.0 mg) were subjected to ODS CC with a gradient of MeOH/H_2_O (from 5:95 to 100:0, v/v), both obtaining ten subfractions, EA6A–EA6J and EA7A–EA7J. Compound 21 (1.3 mg, tR 15.2 min) was obtained from EA6H by semi-preparative HPLC (MeCN–H_2_O, 60:40, v/v, 3 mL/min). Compounds 15 (9.5 mg, tR 20.7 min), 16 (2.0 mg, tR 22.2 min), and 19 (5.5 mg, tR 25.1 min) were purified from EA6I by semi-preparative HPLC (MeCN–H_2_O, 65:35, v/v, 3 mL/min). EA7H was purified by semi-preparative HPLC (MeCN–H_2_O, 60:40, v/v,3 mL/min) to yield 8 (10.9 mg, tR 20.1 min).
Aglodorol A (1): colorless massive crystals; mp 167–168 °C; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{20}_{\text{D}}$$\end{document} + 60.0 (c 0.10, MeOH); UV (MeOH) λmax (logε) 203 nm; IR (KBr) νmax 3320, 2944, 2869, 1643, 1466, 1386, 1377, 1306, 1148, 1044, 1031, 878, 806, 687 cm^− 1^; ^1^H NMR and ^13^C NMR, see Table 1; HRESIMS* m*/z 523.3557 [M + HCOO]^−^ (calcd. for C_31_H_52_O_4_Cl, 523.3554).
Aglodorol B (2), colorless needle crystals; mp 165–167 °C; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{20}_{\text{D}}$$\end{document} + 30 (c 0.10, MeOH); UV (MeOH) λmax (logε) 201 nm; IR (KBr) νmax 3141, 2942, 2871, 1638, 1436, 1405, 1309, 1146, 1114, 1045, 985, 952, 886, 697, 666 cm^− 1^; ^1^H NMR and ^13^C NMR, see Table 1; HRESIMS m/z 489.4301 [M + H]^+^ (calcd. for C_32_H_57_O_3_, 489.4308).
Aglodorol C (8), colorless needle crystals; mp 241–242 °C; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{25}_{\text{D}}$$\end{document} − 90.0 (c 0.10, MeOH); UV (MeOH) λmax (logε) 201 nm; IR (KBr) νmax 3455, 2938, 2872, 1767, 1724, 1461, 1449, 1374, 1249, 1166, 1098, 1034, 979 cm^− 1^; ^1^H NMR and ^13^C NMR, see Table 1; HRESIMS m/z 591.3533 [M + HCOO]^−^ (calcd. for C_33_H_51_O_9_, 591.3528).
Aglodorol D (13), colorless massive crystals; mp 152–153 °C; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{20}_{\text{D}}$$\end{document} − 10.0 (c 0.10, MeOH); UV (MeOH) λmax (logε) 201 nm; IR (KBr) νmax 3326, 2960, 1705, 1467, 1382, 1238, 1168, 1127, 914, 676 cm^− 1^; ^1^H NMR and ^13^C NMR data, see Table 2; HRESIMS* m*/z 305.2480 [M − H]^−^ (calcd. for C_20_H_33_O_2_, 305.2481).
Aglodorol E (14), colorless needle crystals; mp 145–146 °C; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{20}_{\text{D}}$$\end{document} + 60.0 (c 0.10, MeOH); UV (MeOH)* λmax (logε*) 193 nm; IR (KBr) νmax 3385, 2933, 2858, 1706, 1640, 1441, 1380, 1172, 1134, 1027, 1009, 937, 887, 665 cm^− 1^; ^1^H NMR and ^13^C NMR data, see Table 2; HRESIMS* m*/z 305.2480 [M − H]^−^ (calcd. for C_20_H_33_O_2_, 305.2481).
Aglodorol F (15), colorless solid; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{25}_{\text{D}}$$\end{document} + 50 (c 0.10, MeOH); UV (MeOH) λmax (logε) 201 nm; IR (KBr) νmax 3405, 2959, 2869, 1717, 1635, 1469, 1450, 1377, 1303, 1126, 937, 885 cm^− 1^; ^1^H NMR and ^13^C NMR, see Table 2; HRESIMS m/z 351.2539 [M + HCOO]^−^ (calcd. for C_21_H_35_O_4_, 351.2530).
Aglodorol G (16), colorless solid; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{20}_{\text{D}}$$\end{document} + 40 (c 0.20, MeOH); UV (MeOH) λmax (logε) 201 nm; IR (KBr) νmax 3089, 2963, 2932, 2872, 1455, 1375 cm^− 1^; ^1^H NMR and ^13^C NMR, see Table 2; HRESIMS m/z 351.2536 [M + HCOO]^−^ (calcd. for C_21_H_35_O_4_, 351.2541).
Aglodorol H (19), colorless oil; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{25}_{\text{D}}$$\end{document} + 10 (c 0.10, MeOH); UV (MeOH) λmax (logε) 201 nm; IR (KBr) νmax 3425, 2938, 2874, 1710, 1663, 1452, 1375, 1172, 1042, 935 cm^− 1^; ^1^H NMR and ^13^C NMR, see Table 2; HRESIMS m/z 305.2487 [M − H]^−^ (calcd. for C_20_H_33_O_2_, 305.2486).
Aglodorol I (20), light yellow oil; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{20}_{\text{D}}$$\end{document} + 100 (c 0.10, MeOH); UV (MeOH) λmax (logε) 195 nm; IR (KBr) νmax 3500, 2933, 1665, 1619, 1443, 1379, 1183, 1088 cm^− 1^; ^1^H NMR and ^13^C NMR, see Table 1; HRESIMS m/z 303.2327 [M − H]^−^ (calcd. for C_20_H_31_O_2_, 303.2324).
Aglodorol J (21), colorless oil; \documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$[\alpha]^{25}_{\text{D}}$$\end{document} − 20 (c 0.10, MeOH); UV (MeOH) λmax (logε) 201 nm; IR (KBr) νmax 3396, 3078, 2932, 1640, 1613, 1510, 1454, 1377, 1255, 1148 cm^− 1^; ^1^H NMR and ^13^C NMR, see Table 2; HRESIMS m/z 303.2325 [M − H]^−^ (calcd. for C_20_H_31_O_2_, 303.2330).
Crystallographic data and X-ray structure analysis of compounds
Compounds 1, 2, 8, and 13 were obtained as colorless crystals in CHCl_3_, and compound 14 was obtained as colorless crystals in MeOH/H_2_O (1:1, v/v). The crystallographic data for the structures of these compounds have been deposited in the Cambridge Crystallographic Data Centre (deposition Nos.: CCDC 2441755, 2441756, 2441759, 2092476 and 2092472). These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif and Table S1.
Quantum chemistry calculations
The conformation searches of compounds were performed in Spartan 14 with MMFF force field. The primary conformations were then optimized by using DFT method at the B3LYP/6-311G(d) level. The theoretical calculation of NMR was conducted by GIAO method at the mPW1PW91/6-311+G (d, p) in chloroform with PCM model [5]. The calculated NMR data of these conformers were then averaged on the basis of Boltzmann distribution and their relative Gibbs free energies. Subsequent linear regression analysis between experimental and theoretical NMR values yielded a determination coefficient (R^2^) [22]. The ECD calculations were performed at B3LYP/6-311+G(d) level in methanol (PCM model). The calculation ECD curves were fitted in SpecDis v1.71 program with 0.3 eV as the half-bandwidth [23]. All theoretical calculations were implemented in Gaussian 16 [24].
OGD/R model in PC 12 cells
PC 12 cells were plated in 96-well microplates at a density of 7 × 10^3^ cells/well and cultured for 24 h. Following medium removal, the cells underwent gentle PBS washing before exposure to 50 μL EBSS solution containing isolates (10 μM) under anaerobic conditions. After 6 h, each well was replenished with 150 μL complete medium maintaining equivalent drug concentrations for additional 18 h normoxic incubation. The cells in control group were maintained under normoxic conditions throughout the experimental timeline [4]. The cell viability was evaluated by MTT assay, with edaravone (Eda, 16 μM) serving as the positive control.
LPS-induced neuroinflammatory model in BV-2 cells
BV-2 cells (5 × 10^4^ cells/well) were seeded in 48-well plates and cultured overnight. Subsequently, the cells were incubated with LPS (1 μg/mL) and the tested compounds (20 μM) for 24 h. 50 μL of conditioned medium from each well was collected, mixed with Griess reagent (1:1, v/v) and monitored at 540 nm to calculate NO release [5]. Concurrent cytotoxicity assessment was performed via MTT assay. Dexamethasone (Dex, 10 μM) served as the positive drug.
L-glutamate-induced neurotoxicity model in HT22 cells
HT22 cells were plated in 96-well plates at a density of 5 × 10^3^ cells per well and cultured overnight. Following 6 h pre-incubation with the isolates (10 μM), cells were subjected to co-treatment with maintained equivalent compound concentrations and L-glutamic acid (12 mM) in fresh complete medium for 24 h [25]. Cellular viability was quantified via MTT assay.
RSL3-induced ferroptosis in PC12 cells
PC12 cells were seeded in 96-well microplates at a density of 7 × 10^3^ cells/well and cultured for 24 h. Cells were subsequently exposed to ferroptosis inducer RSL3 (0.4 μM) co-administered with the isolates (10 μM) for 4 h [26]. Cell viability was assessed by MTT assay. The ferroptosis inhibitor Fer-1 (10 μM) served as positive control.
Statistical analysis
All results were validated by at least three independent experiments and at least three parallel groups were set up in each experiment. The values were expressed as means ± standard deviation (SD). Statistical analyses were conducted using GraphPad Prism 9.0 software and comparisons were made by independent* t*-tests or one-way analysis of variance (ANOVA). A P-value of < 0.05 was considered statistically significant throughout the study.
Supplementary Information
Additional file 1. X-ray crystallographic data of compounds 1, 2, 8, 13, and 14; HRESIMS, IR, UV, ECD, 1D, 2D NMR spectra of compounds 1–2, 8, 13–16, and 19–21; and the experimental and calculated ECD spectra of compounds 17–18 and 22.
The reference list from the paper itself. Each links out to its DOI / PubMed record.