Transcriptional rewiring in cancer driven by POLR2A/RPB1: mechanistic insights, non-coding RNA crosstalk, and therapeutic opportunities
Adrian Szczepaniak, Kinga Jaskuła, Marta Zielińska, Jakub Godlewski

TL;DR
This paper explores how POLR2A/RPB1, a key component of RNA polymerase II, drives cancer through transcriptional changes and non-coding RNA interactions, offering new therapeutic strategies.
Contribution
The paper reveals novel mechanistic roles of POLR2A/RPB1 in transcriptional rewiring and non-coding RNA crosstalk in cancer.
Findings
POLR2A/RPB1 dysregulation promotes oncogenesis through transcriptional reprogramming and evasion of apoptosis.
Circular POLR2A isoforms modulate tumorigenic pathways by acting as miRNA sponges or protein scaffolds.
Triptolide destabilizes RPB1, highlighting transcription-dependent vulnerabilities in drug-resistant cancers.
Abstract
RNA polymerase II, together with its catalytic subunit RPB1 (encoded by POLR2A), forms the core of the eukaryotic transcriptional machinery that drives the synthesis of protein-coding and regulatory RNA transcripts. Accumulating evidence indicates that dysregulation of POLR2A/RPB1 is a critical driver of oncogenesis, promoting uncontrolled proliferation, evasion of apoptosis, and extensive transcriptional reprogramming across multiple malignancies, frequently affected by recurrent 17p deletions co-occurring with major tumor suppressor loss events. Such coordinated genomic alterations create transcriptional dependency that may be exploited therapeutically. Beyond its canonical role in transcription, POLR2A/RPB1 operates within an extensive regulatory network involving non-coding RNAs. Notably, circular RNAs derived from the POLR2A transcript have emerged as stable post-transcriptional…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
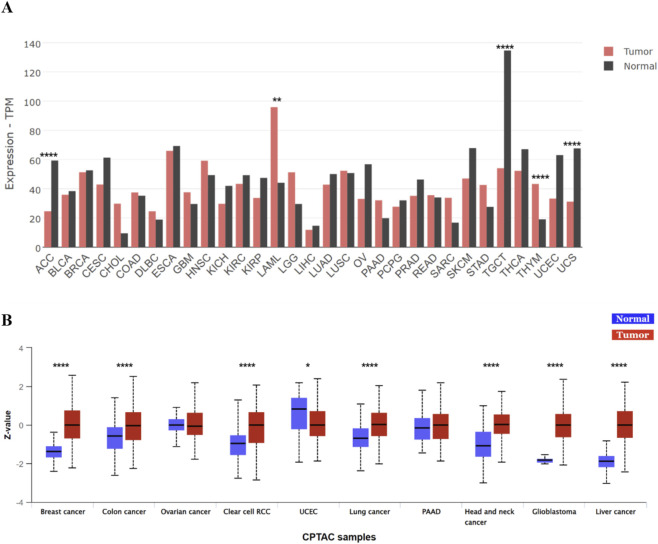 FIGURE 1
FIGURE 1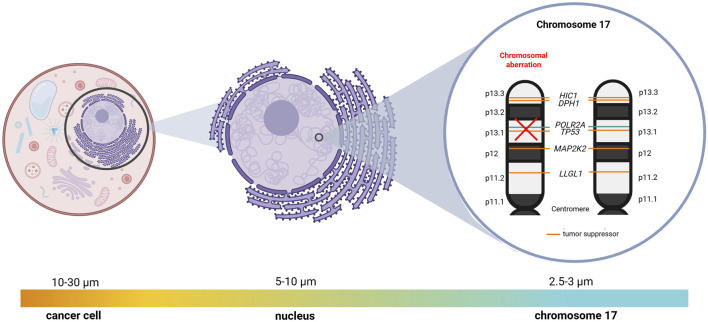 FIGURE 2
FIGURE 2| circAtlas ID | Position | Strand | circRNA type | Multiple conservation score | Spliced length | Algorithm |
|---|---|---|---|---|---|---|
|
| chr17:7495941|7499491 | + | Exonic | 3.0303 | 1578 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7495941|7497045 | + | Exonic | 3.0303 | 726 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7511406|7511588 | + | Exonic | 3.0303 | 183 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7508944|7509190 | + | Exonic | 2.0303 | 247 | find_circ, CIRI2,CIRCexplorer2 |
|
| chr17:7499039|7499491 | + | Exonic | 2.90909 | 336 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7499039|7503791 | + | Exonic | 2.21212 | 131 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7499039|7508475 | + | Exonic | 1.0303 | Unknown | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7503121|7512344 | + | Exonic | 1.06061 | 187 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7501289|7501600 | + | Exonic | 1.0303 | 170 | CIRI2 |
|
| chr17:7499039|7509632 | + | Exonic | 1.09091 | 131 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7511215|7511588 | + | Exonic | 2.0303 | 288 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7503121|7503791 | + | Exonic | 1.06061 | 484 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7497711|7500686 | + | Exonic | 1.0303 | 544 | CIRI2,DCC |
|
| chr17:7503121|7511588 | + | Exonic | 1.09091 | 183 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7513454|7513675 | - | Antisense | 2.0303 | 222 | CIRI2 |
|
| chr17:7514053|7514405 | - | Antisense | 2.0303 | 353 | CIRI2 |
|
| chr17:7508944|7509632 | + | Exonic | 2.06061 | 348 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7501036|7501383 | + | Exonic | 1.0303 | 168 | CIRI2 |
|
| chr17:7500940|7501600 | + | Exonic | 2.0303 | 170 | CIRI2 |
|
| chr17:7508944|7512188 | + | Exonic | 2.0303 | 871 | CIRI2,DCC |
|
| chr17:7497835|7499304 | - | Antisense | 1.0303 | 24 | CIRI2 |
|
| chr17:7501588|7503175 | - | Antisense | 1.0303 | 604 | CIRI2 |
|
| chr17:7501619|7502660 | - | Antisense | 2.0303 | 143 | CIRI2 |
|
| chr17:7500782|7501576 | - | Antisense | 1.0303 | 51 | CIRI2,DCC |
|
| chr17:7501569|7501729 | + | Exonic | 1.0303 | 161 | CIRI2,DCC |
|
| chr17:7508251|7512923 | + | Exonic | 2.06061 | 150 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7503466|7508327 | - | Antisense | 1.0303 | 102 | CIRI2 |
|
| chr17:7484464|7496463 | - | Antisense | 1.0303 | 24 | CIRI2 |
|
| chr17:7503466|7508347 | - | Antisense | 2.0303 | 122 | CIRI2 |
|
| chr17:7503654|7508475 | + | Exonic | 1.0303 | 363 | CIRI2,DCC |
|
| chr17:7509532|7512590 | + | Exonic | 1.0303 | 101 | find_circ, CIRI2,CIRCexplorer2 |
|
| chr17:7503121|7503490 | + | Exonic | 1.0303 | 172 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7502026|7502660 | - | Antisense | 1.0303 | 240 | CIRI2 |
|
| chr17:7499146|7499482 | - | Antisense | 1.0303 | 202 | CIRI2 |
|
| chr17:7500560|7501095 | - | Antisense | 1.0303 | 445 | CIRI2 |
|
| chr17:7503121|7512590 | + | Exonic | 1.0303 | 1850 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7513664|7513833 | - | Antisense | 1.0303 | 170 | CIRI2 |
|
| chr17:7509532|7511319 | + | Exonic | 1.0303 | 206 | find_circ, CIRI2,DCC,CIRCexplorer2 |
|
| chr17:7513517|7513726 | - | Antisense | 3.0303 | 210 | CIRI2 |
|
| chr17:7513732|7513943 | - | Antisense | 2.0303 | 212 | CIRI2 |
|
| chr17:7513664|7513980 | - | Antisense | 3.0303 | 317 | CIRI2 |
|
| chr17:7513876|7514093 | - | Antisense | 3.0303 | 218 | CIRI2 |
|
| chr17:7513454|7513768 | - | Antisense | 2.0303 | 315 | CIRI2 |
|
| chr17:7513652|7513821 | + | Exonic | 2.0303 | 170 | CIRI2 |
|
| chr17:7513786|7513959 | - | Antisense | 2.0303 | 174 | CIRI2 |
|
| chr17:7503466|7503683 | - | Antisense | 2.0303 | 123 | CIRI2 |
|
| chr17:7513454|7513959 | - | Antisense | 2.0303 | 506 | CIRI2 |
|
| chr17:7495941|7503791 | + | Exonic | 1.0303 | Unknown | CIRI2 |
|
| chr17:7513597|7513756 | - | Antisense | 2.0303 | 160 | CIRI2 |
|
| chr17:7513118|7513264 | - | Antisense | 2.0303 | 147 | CIRI2 |
|
| chr17:7513454|7513596 | - | Antisense | 1.0303 | 143 | CIRI2 |
|
| chr17:7513454|7513707 | - | Antisense | 2.0303 | 254 | CIRI2 |
|
| chr17:7513828|7514009 | - | Antisense | 2.0303 | 182 | CIRI2 |
|
| chr17:7513517|7513833 | - | Antisense | 3.0303 | 317 | CIRI2 |
|
| chr17:7513454|7513714 | - | Antisense | 1.0303 | 261 | CIRI2 |
|
| chr17:7513576|7513714 | - | Antisense | 2.0303 | 139 | CIRI2 |
|
| chr17:7509043|7509632 | + | Exonic | 2.0303 | 249 | CIRI2 |
|
| chr17:7496210|7496613 | + | Exonic | 2.0303 | 174 | CIRI2 |
|
| chr17:7513906|7514082 | - | Antisense | 2.0303 | 177 | CIRI2 |
|
| chr17:7512894|7513173 | + | Exonic | 1.0303 | 30 | CIRI2 |
|
| chr17:7497853|7498192 | - | Antisense | 1.0303 | 115 | CIRI2 |
|
| chr17:7503091|7503402 | - | Antisense | 1.0303 | 220 | CIRI2 |
|
| chr17:7513669|7513997 | - | Antisense | 2.0303 | 329 | CIRI2 |
|
| chr17:7513918|7514052 | - | Antisense | 1.0303 | 135 | CIRI2 |
|
| chr17:7498936|7504215 | + | Exonic | 1.0303 | Unknown | CIRI2 |
|
| chr17:7488471|7491379 | + | Non-repeat | 1.0303 | Unknown | CIRI2 |
|
| chr17:7496443|7496581 | - | Antisense | 1.0303 | 139 | CIRI2 |
|
| chr17:7513664|7513875 | - | Antisense | 2.0303 | 212 | CIRI2 |
|
| chr17:7484733|7495962 | + | Exonic | 1.0303 | 147 | CIRI2 |
|
| chr17:7513669|7513850 | - | Antisense | 2.0303 | 182 | CIRI2 |
|
| chr17:7506353|7506510 | + | Non-repeat | 1.0303 | 158 | CIRI2 |
|
| chr17:7512551|7512923 | + | Exonic | 1.0303 | 190 | CIRI2 |
|
| chr17:7484696|7496036 | + | Exonic | 1.0303 | 258 | CIRI2 |
|
| chr17:7513664|7513835 | - | Antisense | 1.0303 | 172 | CIRI2 |
|
| chr17:7509532|7511588 | + | Exonic | 1.0303 | 101 | CIRI2 |
|
| chr17:7498078|7503791 | + | Exonic | 1.0303 | Unknown | CIRI2 |
|
| chr17:7503121|7512923 | + | Exonic | 1.0303 | 156 | CIRI2 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCircular RNAs in diseases · Nuclear Structure and Function · RNA regulation and disease
Introduction
1
The transcriptional machinery of human cells is a finely tuned system that converts the genetic information stored in DNA into functional RNA molecules. At the center of this process is RNA polymerase II (Pol II), the enzyme complex responsible for transcribing all protein-coding genes as well as many non-coding RNA species, including small nuclear RNAs (snRNAs) and microRNAs (miRNAs), which are essential for RNA processing and post-transcriptional regulation (Vlaming et al., 2022). Pol II is composed of 12 subunits, with RPB1, encoded by POLR2A, serving as its most significant and catalytically active core component (Schier and Taatjes, 2020). Proper assembly of these subunits yields a highly efficient transcriptional machine capable of accurately converting the genomic template into messenger RNA. Throughout the transcription cycle - initiation, elongation, and termination - Pol II forms dynamic interactions with transcription factors, mediator complexes, and chromatin regulators that together ensure transcriptional fidelity (Muniz et al., 2021). Because of this central role, Pol II is essential for cell survival and homeostasis.
A hallmark of RPB1 is its C-terminal domain (CTD), composed of multiple tandem repeats of the heptapeptide motif YSPTSPS. Reversible phosphorylation of these repeats coordinates transitions between transcriptional stages and recruits RNA-processing factors required for 5′-capping, splicing, and polyadenylation. These CTD modifications also integrate Pol II function with chromatin remodeling, DNA damage repair, and epigenetic signaling (Yague-Sanz et al., 2023). Comparative studies revealed that RPB1 is highly conserved across eukaryotes; notably, yeast RPB1 can be replaced by its murine ortholog without loss of enzymatic function, underscoring its fundamental role in transcriptional regulation and cellular integrity (Bernecky et al., 2016).
Given its essential function, disruption of POLR2A/RPB1 has widespread consequences. Defects in the complex trigger global transcriptional dysregulation, impaired stress responses, and genomic instability, contributing to various diseases, including cancer (Liu et al., 2015; Li et al., 2018; Xu et al., 2019) and neurodevelopmental disorders (Hansen et al., 2021; Evans et al., 2022). In cancer, POLR2A is frequently overexpressed across tumor types, including breast (Xu et al., 2019), lung (Zeng et al., 2021), colon carcinomas (Liu et al., 2015), and glioblastomas (Wei et al., 2019). Elevated POLR2A levels enhance transcription of genes that support cell proliferation, survival, and metastasis, thereby promoting tumorigenesis. In addition, mutations or aberrations in the regulatory mechanisms–e.g., aberrant CTD phosphorylation–can further destabilize gene expression programs and accelerate cancer progression (Yi et al., 2016).
This mini-review summarizes current knowledge on the role of POLR2A/RPB1 in cancer biology, emphasizing its function as a central Pol II subunit. Integrating both experimental and computational evidence, we discuss how POLR2A dysregulation contributes to tumorigenesis and evaluate its potential as a prognostic biomarker and therapeutic target. We also highlight an emerging perspective on the interplay between POLR2A/RPB1 and non-coding RNAs, particularly circular RNAs derived from the linear POLR2A. These circRNAs may act as stable post-transcriptional regulators that link transcriptional imbalance to oncogenic signaling, opening new avenues for biomarker discovery and targeted therapy.
Distinct POLR2A/RPB1 expression patterns in various cancers
2
A comparative analysis of POLR2A expression across multiple cancer types, conducted using the GEPIA2 platform (Tang et al., 2019), revealed substantial upregulation of POLR2A in malignancies such as acute myeloid leukemia and thymoma. In contrast, strong downregulation was detected in adrenocortical carcinoma, testicular germ cell tumors, and uterine carcinosarcoma (Figure 1A). To complement these transcript-level observations, protein abundance data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) (Li et al., 2023) were analyzed via the UALCAN platform (Chandrashekar et al., 2022) to assess RPB1 expression (Figure 1B). Notably, changes in POLR2A mRNA levels did not always correspond to changes in RPB1 protein levels. For instance, in breast cancer and glioblastoma (GBM), POLR2A levels showed only minor variation, whereas pronounced differences emerged at the protein level (Figures 1A,B). These inconsistencies point to the influence of post-transcriptional regulation, such as changes in mRNA stability, translation efficiency, or protein turnover, that can modulate RPB1 abundance independently of POLR2A transcription.
(A) Expression of POLR2A across various cancers (source: GEPIA2 analyzer). (B) Expression of RPB1 across various cancers (source: UALCAN analyzer). Z-values represent the standard deviation from the median across samples for each cancer type. TPM–Transcripts Per Million; ACC–Adrenocortical carcinoma; BLCA–Bladder Urothelial Carcinoma; BRCA–Breast invasive carcinoma; CESC–Cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL–Cholangio carcinoma; COAD–Colon adenocarcinoma; DLBC–Lymphoid Neoplasm Diffuse Large B-cell Lymphoma; ESCA–Esophageal carcinoma; GBM–Glioblastoma multiforme; HNSC–Head and Neck squamous cell carcinoma; KICH–Kidney Chromophobe; KIRC–Kidney renal clear cell carcinoma; KIRP–Kidney renal papillary cell carcinoma; LAML–Acute Myeloid Leukemia; LGG–Brain Lower Grade Glioma; LIHC–Liver hepatocellular carcinoma; LUAD–Lung adenocarcinoma; LUSC–Lung squamous cell carcinoma; OV–Ovarian serous cystadenocarcinoma; PAAD–Pancreatic adenocarcinoma; PCPG–Pheochromocytoma and Paraganglioma; PRAD–Prostate adenocarcinoma; RCC–Reanl Cell Carcinoma; READ–Rectum adenocarcinoma; SARC–Sarcoma; SKCM–Skin Cutaneous Melanoma; STAD–Stomach adenocarcinoma; TGCT–Testicular Germ Cell Tumors; THCA–Thyroid carcinoma; THYM–Thymoma; UCEC–Uterine Corpus Endometrial Carcinoma; UCS–Uterine Carcinosarcoma. * adjusted p-value <0.05; ** adjusted p-value <0.01; **** adjusted p-value <0.0001.
Taken together, these results demonstrate that POLR2A/RPB1 is broadly dysregulated across cancers, with considerable variability between tumor types. This heterogeneity underscores the importance of further investigation into the regulatory mechanisms governing their expression and the potential of these molecules as biomarkers or therapeutic targets.
Genomic proximity of POLR2A to key tumor suppressor genes: implications for cancer biology and therapy
3
POLR2A, located on chromosome 17p13.1, lies in proximity to the primary tumor suppressor hub - TP53 (Figure 2). Because hemizygous TP53 deletion is common across many cancer types (Yu et al., 2019; van Kampen et al., 2025), POLR2A is frequently co-deleted, resulting in haploinsufficiency of both genes and creating a potential therapeutic vulnerability (Muller et al., 2012; Nijhawan et al., 2012). This genomic relationship has led to the concept of selectively targeting the remaining POLR2A allele to impair tumor cell survival while sparing normal tissues.
Diagram describing co-deletions in POLR2A and TP53 genes (Created with BioRender.com).
In triple-negative breast cancer (TNBC), loss of one POLR2A copy often accompanies TP53 deletion (Xu et al., 2019). Delivery of POLR2A-targeting siRNA via pH-activated nanoparticles effectively suppressed POLR2A-deficient tumors in vivo without systemic toxicity. These nanoparticles accumulated in both POLR2A ^neutral^ and POLR2A ^loss^ tumors, but inhibition of POLR2A significantly reduced tumor growth in heterozygous POLR2A ^loss^ tumors, still avoiding systemic side effects (Xu et al., 2019). A similar pattern is observed in colorectal cancer (CRC), where nearly all TP53-hemizygous loss cases show concurrent POLR2A deletion (Liu et al., 2015). In these models, inhibition of POLR2A by α-amanitin or specific siRNA markedly reduced proliferation and tumor growth in POLR2A ^loss^ models, independently of p53 status, suggesting a p53-independent therapeutic mechanism (Liu et al., 2015). These findings indicate that therapeutic targeting of POLR2A may represent a broadly effective, p53-independent strategy for tumors harboring TP53 deletions.
In castration-resistant prostate cancer (CRPC), 17p deletions occur in approximately 63% of metastatic cases (Li et al., 2018). In this context, RPB1 interacts with the E3 ligase RBX1, which enhances its activity via K63-linked ubiquitination, thereby augmenting RNAP2-mediated mRNA synthesis. Supporting the relevance of this mechanism, TCGA prostate cancer data show an inverse correlation between RBX1 and POLR2A expression (R = −0.45), suggesting that the loss of POLR2A in cells with deletion of the 17p chromosomal region may be functionally compensated by RBX1 upregulation (Li et al., 2018). Consequently, combined inhibition of RNAP2 and RBX1 markedly suppresses CRPC growth through a synergistic effect.
Beyond chromosomal deletions, somatic POLR2A mutations have also been identified in a cohort of 775 meningioma patients lacking known driver alterations; recurrent mutations such as p.Gln403Lys or a deletion of the p.Leu438_His439 region were detected (Clark et al., 2016). These tumors were genomically stable and had a low mutation burden, comparable to meningomas driven by established genes. Functionally, POLR2A mutations disrupted the expression of key meningeal identity genes (WNT6, ZIC1/ZIC4), defining a distinct and clinically unique meningioma subgroup (Clark et al., 2016). Collectively, these findings reinforce the central importance of POLR2A in transcriptional regulation and highlight its therapeutic vulnerability, particularly in cancers with 17p deletions or POLR2A mutations.
Diverse cellular pathways underlying POLR2A-driven oncogenesis
4
POLR2A is markedly upregulated in gastric cancer (GC) tissues compared with adjacent non-tumor tissue, and its elevated expression correlates with larger tumor size, deeper invasion, and poorer prognosis (Jiang et al., 2021). Functional studies revealed that POLR2A knockdown in POLR2A ^high^ cells markedly reduced viability. In contrast, its overexpression in POLR2A ^low^ cells enhanced viability and inhibited apoptosis by upregulating BCL2 and PARP, supporting its role in GC growth both in vitro and in vivo (Jiang et al., 2021). In lung adenocarcinoma, POLR2A expression strongly correlates with BCAR1 (breast cancer antiestrogen resistance protein 1) overexpression and with unfavorable clinical outcomes (Mao et al., 2020). BCAR1 knockout markedly suppressed cell proliferation and colony formation and, significantly, led to a substantial reduction in RPB1 levels. This suggests a regulatory interaction between BCAR1 and RPB1 that may contribute to the aggressive behavior of lung adenocarcinoma (Mao et al., 2020). In acute myeloid leukemia (LAML), both POLR2A and its catalytic subunit RPB1 are aberrantly activated, with RPB1 levels positively correlating with tumor burden and poor outcome (Yu et al., 2019). Global transcriptomic profiling showed that POLR2A knockout markedly suppressed oncogenic and antiapoptotic genes, including MYC, RUNX2, MEIS1, CDC25A, and BCL-2, leading to impaired proliferation and a significant reduction in tumor cell populations at 7 and 14 days after implantation in vivo (Yu et al., 2019).
Beyond changes in expression levels, the subcellular localization of RPB1 also appears to shape tumor behavior and therapy response. Although RPB1 normally resides in the nucleus, cancer cells can form cytoplasmic RPB1 aggregates, particularly in therapy-resistant breast and renal tumors (Nagy-Mikó et al., 2023). These aggregates, absent in normal tissue, likely reflect dysregulated translation as a compensatory response to genotoxic stress. Importantly, their presence in pretreatment biopsies may serve as a predictive marker of chemotherapy resistance (Nagy-Mikó et al., 2023).
Taken together, these findings show that POLR2A and RPB1 contribute to oncogenesis through multiple mechanisms, including regulation of apoptosis, cell cycle progression, and transcription of oncogenic programs. Moreover, both their expression patterns and subcellular localization hold promise as prognostic indicators and therapeutic targets across diverse cancer types.
Targeting RPB1 in cancer therapy: transcriptional vulnerabilities as a therapeutic opportunity
5
Triptolide, a diterpenoid compound originally isolated from Tripterygium wilfordii, has attracted considerable attention for its anticancer activity. It acts on multiple cellular pathways, including transcriptional regulation, apoptosis, cell cycle progression, oxidative stress responses, and autophagy (Bao et al., 2024; Feng et al., 2024). Mechanistic studies by Manzo et al. (Manzo et al., 2012) showed that triptolide selectively induces phosphorylation of the CTD at Ser-5 as an early event, followed by a progressive decrease in total RPB1 levels. Triptolide also caused promoter-proximal stalling of RNA polymerase II and reduced chromatin-bound Pol II, highlighting a direct impact on transcriptional initiation (Manzo et al., 2012). The importance of RPB1 degradation for drug sensitivity was further demonstrated in multidrug-resistant cancer models, where RPB1 was critical for mediating triptolide’s cytotoxic effects (Yi et al., 2016). CDK7-dependent phosphorylation of RPB1 at Thr170 promoted its degradation, while treatment with the selective CDK7 inhibitor BS-181 partially rescued cell viability (Yi et al., 2016). Together, these findings establish RPB1 destabilization as a hallmark of triptolide’s mechanism of action and suggest that RPB1 itself represents a promising therapeutic vulnerability, particularly in cancers resistant to conventional chemotherapies.
Targeting POLR2A/RPB1: viral strategies for transcriptional dominance
6
Many viruses that rely on host transcription have evolved sophisticated mechanisms to manipulate or disable POLR2A/RPB1 to seize control of cellular gene expression. By targeting this essential enzyme, viruses establish transcriptional dominance, boosting viral RNA synthesis while suppressing host antiviral defenses.
RNA viruses such as influenza and bunyaviruses hijack Pol II-derived capped RNA fragments to initiate their own mRNA synthesis. This strategy not only ensures efficient translation of viral transcripts but also helps the virus evade innate immune detection (Ding and Summers, 2022; Xu M. et al., 2022). In contrast, nuclear DNA viruses, including herpesviruses and adenoviruses, reprogram Pol II activity more directly. They modify CTD phosphorylation patterns to suppress host transcription while maintaining robust viral gene expression selectively (Zaborowska et al., 2014; Isa et al., 2021; Huang et al., 2022; Whisnant et al., 2024).
Cytoplasmic viruses, such as alphaviruses, bunyaviruses, and coronaviruses, often deploy a more aggressive approach. They trigger ubiquitin-proteasome-mediated degradation of RPB1, causing a rapid, global shutdown of host mRNA synthesis (Akhrymuk et al., 2012; Schoen et al., 2020; Hardy et al., 2024). Despite their diverse replication strategies, these viruses converge on a common principle: disabling RPB1, either through functional inactivation or degradation, is an efficient way to silence host defenses and monopolize the transcriptional machinery.
This repeated viral targeting of POLR2A/RPB1 highlights its central importance in transcriptional fidelity and cellular stress resilience. Intriguingly, cancer cells display analogous dependencies, exploiting POLR2A/RPB1 dysregulation to sustain aberrant transcriptional programs and survive genotoxic stress. Recognizing this functional overlap between viral infection and tumor biology opens new therapeutic opportunities: oncolytic viruses and transcription-targeted agents may be strategically leveraged to disrupt tumor-specific vulnerabilities driven by the POLR2A/RPB1 axis.
Deciphering the regulatory nexus: crosstalk between POLR2A and non-coding RNAs
7
Growing evidence indicates that non-coding RNAs (ncRNAs) – once regarded as the “dark matter” of the genome–are key regulators of cancer development and progression (Guzel et al., 2019; Duan et al., 2023). Although they do not encode proteins, ncRNAs are highly abundant: over 98% of the human genome is non-coding, and roughly 93% of these sequences are actively transcribed (ENCODE Project Consortium, 2012; Wang, 2022). The ncRNA family comprises several subclasses, including miRNAs, long non-coding RNAs (lncRNAs), and the more recently identified circular RNAs (circRNAs) (Szczepaniak et al., 2023). Emerging studies link POLR2A to multiple aspects of ncRNA biology, showing that it regulates lncRNA expression and also serves as a transcriptional template for circRNA biogenesis, suggesting a broad influence on the non-coding transcriptome.
In the canonical model, lncRNAs are transcribed by Pol II and regulated by various protein factors (Statello et al., 2021; Mattick et al., 2023). However, large-scale analyses have proposed additional, noncanonical modes of lncRNA transcription, potentially involving unknown regulators or lncRNA-lncRNA interactions (Wang, 2022). Intriguingly, lncRNAs interact with molecular effectors–such as transcription factors, histone modifications, or other regulatory proteins–in patterns that are noticeably different from those seen with protein-coding genes. Only ∼12% of lncRNA promoter regions (defined as ±5 kb around the transcription start site) are predicted to bind RPB1 (Wang, 2022), challenging the long-standing assumption that Pol II is the primary driver of all lncRNA transcription. Several studies have specifically implicated POLR2A-associated lncRNAs in cancer. In ovarian cancer, Guo et al., 2019) demonstrated that the signature of POLR2A-associated lncRNAs is more sensitive and specific for predicting survival than current clinical and molecular markers. Their findings showed that the POLR2A-lncRNA signature effectively stratified patients into high- and low-risk groups based on overall, progression-free, and disease-free survival. Furthermore, the signature effectively divided patients into distinct risk subgroups across various clinical parameters, including age, tumor grade, stage, and residual tumor diameter (Guo et al., 2019). These findings highlight the potential of POLR2A-linked lncRNAs as robust prognostic biomarkers and possible therapeutic targets.
Beyond its regulatory role in linear ncRNA expression, POLR2A also serves as a template for circRNA production (Chen et al., 2022; Xu Z. et al., 2022). CircRNAs are generated through a unique back-splicing process, in which a downstream 5′splice site covalently joins an upstream 3′splice site, developing a closed-loop structure devoid of free ends. This circular structure confers exceptional stability and resistance to exonuclease degradation (Yu and Kuo, 2019). Functionally, circRNAs act as miRNA sponges, serve as scaffolds for protein interactions, modulate transcription, and, in some cases, can even be translated (Verduci et al., 2021). Increasingly, circRNAs have been linked to oncogenic signaling and are emerging as promising biomarkers and therapeutic targets (Bronisz et al., 2020; Shi et al., 2024).
According to circBase, at least 77 POLR2A-derived circRNAs have been identified (Table 1) (Glažar et al., 2014). Among them, circPOLR2A (hsa-POLR2A_0005) is the most extensively studied. This circRNA is strongly upregulated in clear cell renal cell carcinoma (ccRCC), including metastatic lesions, and promotes proliferation, migration, invasion, and angiogenesis while inhibiting apoptosis (Xu Z. et al., 2022). Mechanistically, hsa-POLR2A_0005 forms a ternary complex with UBE3C and PEBP1, thereby enhancing PEBP1 ubiquitination and degradation, activating the ERK signaling pathway, and driving tumor progression. In addition, m^6^A modification of hsa-POLR2A_0005 modulates its stability and function, further influencing ccRCC pathogenesis. Targeting the *hsa-POLR2A_0005/*UBE3C/PEBP1 axis, therefore, represents a potential therapeutic approach for this aggressive malignancy (Xu Z. et al., 2022). * hsa-POLR2A_0005
- is also upregulated in GBM (Chen et al., 2022), where it promotes proliferation and inhibits apoptosis. Subcellular fractionation revealed that hsa-POLR2A_0005 is predominantly cytoplasmic and acts as a sponge for miR-2113. By sequestering miR-2113, * hsa-POLR2A_0005
- upregulates POU3F2, thereby activating SOX9 transcription through direct interaction with its promoter. These findings reveal a novel mechanistic pathway driving GBM progression, suggesting that targeting the hsa-POLR2A_0005 miR-2113/POU3F2/SOX9 axis could offer valuable therapeutic opportunities (Chen et al., 2022). Although hsa-POLR2A_0005 is the best characterized, many other circRNAs derived from the POLR2A locus likely participate in transcriptional or post-transcriptional regulation and remain to be explored.
CircRNAs can also modulate POLR2A activity themselves. In ovarian cancer, circMETTL6 has been identified as a tumor suppressor, where its overexpression inhibits metastatic growth and prolongs survival (Yu et al., 2025). Mechanistic studies revealed that loss of circMETTL6 increases NONO expression and stabilizes RPB1, promoting GDF15 transcription and metastatic potential (Yu et al., 2025). These results suggest that circRNAs derived from other genomic loci can influence POLR2A/RPB1-dependent transcriptional programs in cancer.
Regulation of POLR2A may also involve other classes of ncRNAs, including miRNAs. miRNAs primarily control gene expression by binding to target mRNAs in the cytoplasm, which can result in mRNA degradation and subsequent recycling of its components or in preserving the mRNA for later translation (O’Brien et al., 2018). Depending on the context, their dysregulation can contribute to tumorigenesis, acting as oncogenes or tumor suppressors. Altered miRNA expression profiles have been linked to key processes such as regulation of molecular landscape, proliferation, apoptosis, and metastasis, making them valuable biomarkers for cancer diagnosis and prognosis (Rooj et al., 2017; Metcalf, 2024). Furthermore, therapeutic strategies targeting miRNAs are being explored, offering promising avenues for personalized cancer treatment (Kim and Croce, 2023). The role for miR-378a-3p was discovered in sarcomatoid renal cell carcinoma (sRCC) using clinically derived cell lines. The authors showed that the mRNA and protein expression levels of POLR2A and RUNX2 were significantly higher than in other RCC subtypes. Mechanistically, increased levels of miR-378a-3p expression in sRCC cell lines (RCC52) suppressed RPB1 and RUNX2 and enhanced apoptosis (Weng et al., 2022). These findings highlight the possible intricate post-transcriptional regulation of POLR2A by a diverse array of miRNAs, underscoring their potential as modulators of oncogenic pathways.
Conclusion and future directions
8
Recent results have underscored the pivotal role of POLR2A/RPB1 dysregulation in driving tumor growth and progression across various cancer types. Elevated POLR2A expression is consistently associated with increased cell proliferation, altered cell-cycle dynamics, and resistance to apoptosis, highlighting its importance in sustaining oncogenic programs. Moreover, studies suggest that POLR2A/RPB1 interacts with various proteins to form regulatory networks that may drive aggressive tumor behavior and affect treatment responses.
Future efforts should focus on dissecting the complex molecular crosstalk between POLR2A and key cellular regulators. Integrative approaches, combining multi-omics profiling, functional genomics, epitranscriptomics, and advanced computational modeling, will be crucial for fully mapping these networks and identifying new therapeutic vulnerabilities.
The promising anticancer potential of natural compounds warrants further exploration as they may offer innovative strategies to modulate POLR2A/RPB1 activity and combat drug resistance. Bioactive compounds such as triptolide have already demonstrated the ability to disrupt transcriptional machinery. Future research should focus on elucidating the precise molecular mechanisms by which these compounds regulate POLR2A and its associated pathways, potentially unveiling new therapeutic targets. Moreover, pairing natural compounds with advanced drug-delivery platforms, including nanoparticle-based systems, may enhance their stability, specificity, and clinical efficacy.
The recurrent targeting of POLR2A/RPB1 by phylogenetically diverse viruses further highlights its role as a nonredundant bottleneck within the cellular transcriptional apparatus. Through mechanisms such as cap-snatching, CTD phosphorylation rewiring, and proteasome-mediated RPB1 degradation, viruses exploit Pol II to silence host transcription and enforce viral gene expression. The CTD code, once viewed as rigid, is now understood as a dynamic regulatory interface that viruses can reprogram to establish noncanonical transcriptional states. Intriguingly, similar principles appear to operate in cancer, where POLR2A/RPB1 dysregulation fuels transcriptional addiction and malignant progression. These parallels suggest a unifying conceptual framework: understanding RPB1 vulnerabilities may inspire therapeutic innovation spanning transcription-targeted agents to rationally engineered oncolytic viruses. Although still speculative, viruses such as HSV-1 could potentially be modified to exploit Pol II activity selectively within tumors, dismantling cancer-specific transcriptional programs while amplifying viral oncolysis.
Another emerging dimension of POLR2A-driven oncogenesis is the production of circRNAs derived from the POLR2A transcript. CircRNAs are increasingly recognized as important regulators of gene expression and cancer biology. Continued research into the circularization of the pre-POLR2A transcript may clarify how these molecules contribute to tumor progression and could lead to the development of new diagnostic tools and therapeutic strategies. Because POLR2A is a protein-coding gene, identifying potential open reading frames within circPOLR2A isoforms may also shed light on noncanonical translation events in cancer. High-throughput sequencing will be essential for cataloging the full repertoire of circPOLR2As, ultimately enabling the discovery of novel biomarkers and actionable therapeutic targets.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Akhrymuk I. Kulemzin S. V. Frolova E. I. (2012). Evasion of the innate immune response: the old world alphavirus ns P 2 protein induces rapid degradation of Rpb 1, a catalytic subunit of RNA polymerase II. J. Virol. 86, 7180–7191. 10.1128/JVI.00541-12 22514352 PMC 3416352 · doi ↗ · pubmed ↗
- 2Bao S. Yi M. Xiang B. Chen P. (2024). Antitumor mechanisms and future clinical applications of the natural product triptolide. Cancer Cell Int. 24, 150. 10.1186/s 12935-024-03336-y 38678240 PMC 11055311 · doi ↗ · pubmed ↗
- 3Bernecky C. Herzog F. Baumeister W. Plitzko J. M. Cramer P. (2016). Structure of transcribing mammalian RNA polymerase II. Nature 529, 551–554. 10.1038/nature 16482 26789250 · doi ↗ · pubmed ↗
- 4Bronisz A. Rooj A. K. Krawczyński K. Peruzzi P. Salińska E. Nakano I. (2020). The nuclear DICER-circular RNA complex drives the deregulation of the glioblastoma cell micro RN Aome. Sci. Adv. 6, eabc 0221. 10.1126/sciadv.abc 0221 33328224 PMC 7744081 · doi ↗ · pubmed ↗
- 5Chandrashekar D. S. Karthikeyan S. K. Korla P. K. Patel H. Shovon A. R. Athar M. (2022). UALCAN: an update to the integrated cancer data analysis platform. Neoplasia 25, 18–27. 10.1016/j.neo.2022.01.001 35078134 PMC 8788199 · doi ↗ · pubmed ↗
- 6Chen Z. Mai Q. Wang Q. Gou Q. Shi F. Mo Z. (2022). Circ POLR 2A promotes proliferation and impedes apoptosis of glioblastoma multiforme cells by Up-regulating POU 3F 2 to facilitate SOX 9 transcription. Neuroscience 503, 118–130. 10.1016/j.neuroscience.2022.03.035 35398178 · doi ↗ · pubmed ↗
- 7Clark V. E. HarmancıA. S. Bai H. Youngblood M. W. Lee T. I. Baranoski J. F. (2016). Recurrent somatic mutations in POLR 2A define a distinct subset of meningiomas. Nat. Genet. 48, 1253–1259. 10.1038/ng.3651 27548314 PMC 5114141 · doi ↗ · pubmed ↗
- 8Ding P. Summers M. F. (2022). Sequestering the 5′-Cap for viral RNA packaging. Bioessays 44, e 2200104. 10.1002/bies.202200104 36101513 PMC 10201543 · doi ↗ · pubmed ↗