Epithelial to Mesenchymal Transition Transcriptional Regulator ZEB1 in Liver Cancer: Oncogenic Roles and Therapeutic Potential
Ester Gonzalez-Sanchez, Carlos Andres Roldan-Hernandez, Ana Martin-Ramirez, Lucia Garcia-Collado, Laura Fouassier, Javier Vaquero

TL;DR
This paper explores how ZEB1, a key regulator of cell transition, contributes to liver cancer and how targeting it could lead to better treatments.
Contribution
The paper provides new insights into ZEB1's role in liver cancer progression and its potential as a therapeutic target.
Findings
ZEB1 is overexpressed in liver tumors like HCC and CCA.
High ZEB1 levels correlate with advanced disease and poor survival in liver cancer patients.
Abstract
Zinc finger E-box binding homeobox 1 (ZEB1) is a member of the zinc finger homeodomain transcription factor family, with a pivotal role in regulating the epithelial to mesenchymal transition (EMT) process. Increasing evidence suggests that ZEB1 is overexpressed in liver tumors, including hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA), and it correlates with advanced disease features and reduced overall survival. Here, we examine ZEB1 molecular functions, regulatory networks and contribution to tumorigenesis. We also discuss the emerging therapeutic strategies and future research directions aimed at targeting the ZEB1 molecular network to improve the outcome of liver cancer patients.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
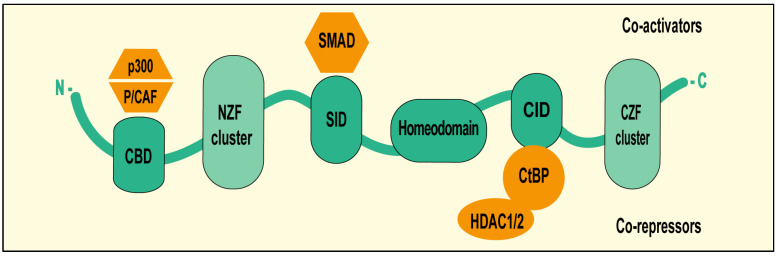 Figure 1
Figure 1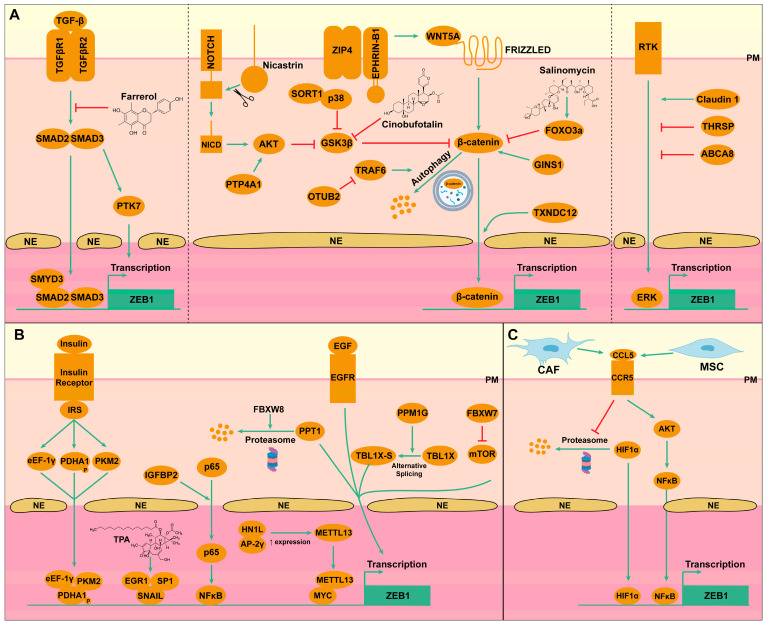 Figure 2
Figure 2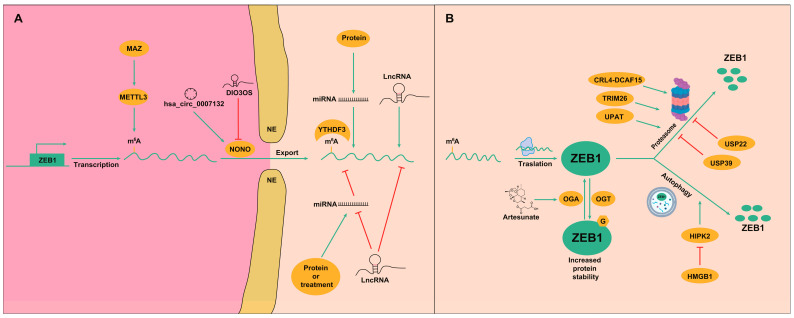 Figure 3
Figure 3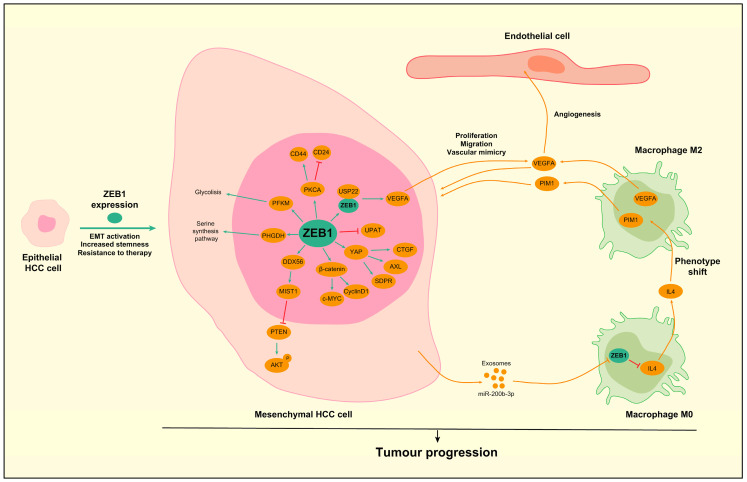 Figure 4
Figure 4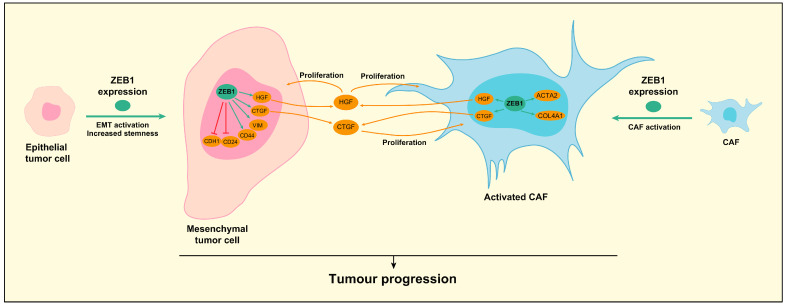 Figure 5
Figure 5| Tumor | Factor | Regulates | miRNA | Regulates | Factor | Result | Ref. |
|---|---|---|---|---|---|---|---|
| HCC | Lnc-RP11-422N16.3 | Negatively | miR-23b-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | LncRNA-SNHG6 | Negatively | miR-101-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | - | - | miR-139-5p | Negatively | ZEB1 | ↓ ZEB1 | [ |
| HCC | LncRNA-TUG1 | Negatively | miR-142-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | Lnc-MALAT1 | Negatively | miR-143-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | circ-100338 | Negatively | miR-143-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | ELF3 | Negatively | miR-141-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | ATRA | Positively | miR-141-3p | Negatively | ZEB1 | ↓ ZEB1 | [ |
| HCC | LncRNA-ZFAS1 | Negatively | miR-150 | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | TGF-β | Positively | miR-155 | Positively | ZEB1 | ↑ ZEB1 | [ |
| HCC | LncRNE-PE | Negatively | miR-200a | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | Gα12 | Negatively | miR-200a | Negatively | ZEB1 | ↑ ZEB1 | [ |
| CCA | Lnc-ZEB1-AS1 | Negatively | miR-200a | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | Lnc-HULC | Negatively | miR-200a-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | Genomic deletion, promoter methylation | Negatively | miR-200b | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | Lnc-CARLo-5 | Negatively | miR-200b | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | KLF4 | Positively | miR-200b | Negatively | ZEB1 | ↓ ZEB1 | [ |
| HCC | YB1 | Negatively | miR-200b | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | LncRNA-XIST | Negatively | miR-200b-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| CCA | - | - | miR-200c | Negatively | ZEB1 | ↓ ZEB1 | [ |
| CCA | Lnc-ATB | Negatively | miR-200c | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | p53 | Positively | miR-200 | Negatively | ZEB1 | ↓ ZEB1 | [ |
| HCC | LncRNA-ATB | Negatively | miR-200 | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | LINC00273 | Negatively | miR-200 | Negatively | ZEB1 | ↑ ZEB1 | [ |
| CCA | Lnc-NNT-AS1 | Negatively | miR-203 | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | Lnc-SNHG3 | Negatively | miR-326 | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | - | - | miR-369 | Negatively | ZEB1 | ↓ ZEB1 | [ |
| HCC | LncRNA-PRNCR1 | Negatively | miR-411-3p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | Lnc-MAPKAPK5 | Negatively | miR-429 | Negatively | ZEB1 | ↑ ZEB1 | [ |
| CCA | DMY | Positively | miR-455-3p | Negatively | ZEB1 | ↓ ZEB1 | [ |
| HCC | - | - | miR-590-3p | Negatively | ZEB1 | ↓ ZEB1 | [ |
| HCC | - | - | miR-708 | Negatively | ZEB1 | ↓ ZEB1 | [ |
| HCC | Lnc171 | Negatively | miR-873-5p | Negatively | ZEB1 | ↑ ZEB1 | [ |
| HCC | Lnc-HCCL5 | - | - | Positively | ZEB1 | ↑ ZEB1 | [ |
| HCC | LncPNUTS | - | - | Positively | ZEB1 | ↑ ZEB1 | [ |
- —Agencia Estatal de Investigación (AEI), Ministry of Science and Innovation (MICIN), Spain
- —FEDER funds/Development Fund—a way to build Europe
- —Fundación Memoria de D. Samuel Solorzano Barruso
- —«Escalera de Excelencia» of the Education Ministry of the Castilla y León autonomous government
- —Ramon y Cajal
- —CSIC through a JAE-PRE fellowship
- —Instituto de Salud Carlos III, Spain
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer Cells and Metastasis · Developmental Biology and Gene Regulation · Liver physiology and pathology
1. Introduction
Liver cancer includes hepatocellular carcinoma (HCC), which is the most prevalent form of primary liver cancer and accounts for approximately 75–85% of cases [1], and cholangiocarcinoma (CCA), which arises from the epithelial cells that line the bile ducts, and represents around 10–15% of all liver cancer cases [2]. Despite fundamental differences in etiological factors, genetic drivers and tumor microenvironment (TME) characteristics, both HCC and CCA are characterized by aggressive behavior, poor prognosis and limited therapeutic options, especially in the advanced stages of the disease, which occur in the majority of patients due to frequent late-stage diagnosis of these malignancies [1,2]. Consequently, liver cancer currently ranks as the third leading cause of cancer-related mortality [3].
Among the mechanisms leading to cell dissemination and metastasis of cancer cells, the epithelial to mesenchymal transition (EMT) is probably the most widely studied mechanism in this regard [4]. EMT, originally observed during embryogenesis, is a reversible dynamic process during which epithelial cells gradually adopt the structural and functional characteristics of mesenchymal cells, including motility, invasiveness, stemness and chemoresistance [4,5]. This process is tightly regulated by different environmental signals from major signaling pathways (i.e., transforming growth factor-beta (TGF-β), Fibroblast growth factor (FGF), wingless/integrated (Wnt), etc.) and a group of EMT-inducing transcription factors (EMT-TF) belonging to three main families (zinc finger E-box-binding homeobox 1/2 (ZEB1/2), snail family transcriptional repressor 1/2 (SNAI1/2) and twist family bHLH transcription factor 1/2 (TWIST1/2)) [4,5]. These factors, mainly identified as negative regulators of epithelial markers such as E-cadherin, have been involved in a plethora of molecular mechanisms involved in various biological processes since then [6,7,8].
The ZEB (Zinc finger E-box-binding homeobox) protein family of transcription factors was first discovered in Drosophila melanogaster and is constituted by two members, ZEB1 and ZEB2 [9]. Both of them share structural similarities and contain two zinc finger clusters that allow them to bind DNA regulatory sequences and, therefore, control the expression of their targets through promoter binding [9]. ZEB1 and ZEB2 are known for their ability to stimulate EMT, but some reports indicate that they can have both redundant and opposing functions, even in the same type of cancer [9,10,11].
The most studied ZEB protein is undoubtedly ZEB1, which has been shown to play multiple roles in regulating the aggressivity and chemoresistance of different tumor types [12]. Emerging evidence suggests that ZEB1 is overexpressed in liver tumors compared to normal liver tissue, and that its expression correlates with advanced disease features such as vascular invasion, intrahepatic metastasis and reduced overall survival [13,14]. Thus, understanding ZEB1-related functions in liver cancer may introduce new ways to therapeutically challenge these aggressive malignancies.
In this review, we aim to give a comprehensive overview of the current knowledge on the biological roles of ZEB1 in the two main subtypes of liver cancer, HCC and CCA. We will first introduce the structural and functional characteristics of ZEB1, followed by a detailed examination of its role in tumor initiation, progression and resistance mechanisms in both types of liver cancer. Finally, we will discuss emerging therapeutic strategies and future directions for research, aimed at exploiting ZEB1 as a biomarker and therapeutic target.
2. ZEB1 Structural Features and Functional Domains
The ZEB1 protein (also known as TCF8 and δEF1) is encoded by the homonym gene located on chromosome 10p11.22 [15]. The protein consists of 1117 amino acids and is a transcription factor that features two clusters of C2H2-type zinc finger domains at both the N-terminal and C-terminal regions, which are responsible for binding to the E-box sequences in target gene promoters with a consensus sequence of 5′-CANNTG-3′ (Figure 1) [16]. The middle region contains a homeodomain that contributes to protein–protein interactions. In addition, ZEB1 contains a Smad interaction domain (SID), a CtBP interaction domain (CID) and p300-P/CBP-associated factor (P/CAF) binding domain (CBD) that are key in the control of its transcriptional activity (Figure 1) [17]. ZEB1 may upregulate or downregulate target gene transcription through the recruitment of different coactivators or cosuppressors via SID, CID and CBD [17]. For example, ZEB1 can directly interact with SMADs through SID to regulate TGF-β-responsive genes. Similarly, p300 and P/CAF can be recruited to CBD to activate gene transcription. Conversely, ZEB1 can repress target gene transcription through the recruitment of the CtBP corepressor complex (histone deacetylases HDAC1/2) via CID (Figure 1) [18]. These cofactors allow ZEB1 to modulate transcription with epigenetic mechanisms, including DNA methylation or histone modifications [19,20]. This extensive list of partners allows ZEB1 to play pleiotropic functions in various biological processes, from embryogenesis to cancer.
3. Regulation of ZEB1 Expression in Liver Cancer
3.1. Transcriptional Regulation
ZEB1 is not only a key effector of EMT but is also integrated into complex regulatory networks involving major signaling pathways, which are frequently dysregulated in liver cancers. The mechanisms of regulation of ZEB1 transcription are summarized in Figure 2. For example, TGF-β is able to upregulate ZEB1 expression in HCC cells in a SMAD2/3-dependent mechanism (Figure 2A) [21,22]. On one side, SMAD2/3 are able to upregulate the expression of Tyrosine-protein kinase-like 7 (PTK7), which in turn upregulates ZEB1 expression. On the other side SMAD2/3 are able to directly bind the ZEB1 promoter to promote ZEB1 transcription [21]. Interestingly, SET (suppressor of variegation, enhancer of zeste, trithorax) and MYND (Myeloid-Nervy-DEAF1) domain containing 3 (SMYD3) is required for TGF-β-induced EMT in HCC cells that are resistant to sorafenib. Indeed, SMYD3 interacts with SMAD2/3 and epigenetically promotes the expression of ZEB1 [21].
Another major signaling cascade involved in ZEB1 regulation is the Wnt/β-catenin pathway (Figure 2A). Generally, WNT signaling activation disrupts the binding of Glycogen synthase kinase 3 beta (GSK3β) to β-catenin to allow the later to translocate to the nucleus and activate target gene transduction, including ZEB1, but several other molecules can modulate this cascade in liver cancer. For example, upstream WNT, Zrt- and Irt-like protein 4 (ZIP4), a zinc transporter located at the plasma membrane, inhibits by direct binding the ubiquitination of Ephrin-B1, which in turn activates WNT5A signaling in HCC cells [23]. Downstream of WNT, several mechanisms also regulate the activity of β-catenin in HCC cells. A thioredoxin domain containing 12 (TXNDC12) interacts with β-catenin and stimulates the nuclear translocation and activation of β-catenin [24]. Nicastrin (NCSTN) also facilitates the translocation of β-catenin to the nucleus. More precisely, Nicastrin activates Notch1 cleavage, which in turn activates AKT serine/threonine kinase (AKT) signaling, leading to GSK3β phosphorylation and liberation of β-catenin from the GSK3β/β-catenin complex [25]. Similarly, Sortilin 1 (SORT1) is able to bind p38 to enhance its stability and promote GSK-3β phosphorylation and the liberation of β-catenin [26]. Other factors affect β-catenin signaling in HCC cells. GINS complex subunit 1 (GINS1) promotes EMT and tumor metastasis by favoring β-catenin signaling [27], while Forkhead box O3 (FOXO3a) reduces the binding of β-catenin to the T-cell factor (TCF) and inhibits β-catenin/TCF target gene expression [28]. Although less studied, some other mechanisms regulating β-catenin have been identified in CCA. For example, the ubiquitin thioesterase OTU deubiquitinase, ubiquitin aldehyde binding 2 (OTUB2), is able to inhibit the lysosomal degradation of β-catenin by interacting with the TNF receptor-associated factor 6 (TRAF6), resulting in increased expression and signaling of β-catenin [29]. Another mechanism involves Protein tyrosine phosphatase type 4A1 (PTP4A1), which activates AKT signaling, which in turn phosphorylates GSK3β, liberating β-catenin to translocate to the nucleus and activate its target gene transcription [30].
Another ZEB1 major regulatory cascade is the Mitogen-activated protein kinase (MAPK) signaling pathway (Figure 2A). Activation of different receptors leads to the activation of this pathway, which ends with the translocation of phosphorylated extracellular signal-regulated kinase (ERK) to the nucleus and its binding to the ZEB1 promoter. In HCC cells, the thyroid hormone responsive protein (THRSP) and ATP binding cassette subfamily A member 8 (ABCA8) inhibit ERK signaling and ZEB1-induced EMT [31,32]. On the contrary, claudin-1 activates the MAPK pathway and ZEB1 expression [33].
Other signals can also regulate ZEB1 expression (Figure 2B). In HCC cells, insulin binding to the insulin receptor triggers the phosphorylation of pyruvate dehydrogenase E1 subunit alpha 1 (PDHA-1) and its nuclear translocation in the company of eukaryotic translation elongation factor 1 gamma (eEF-1γ) and pyruvate kinase M2 (PKM2), which together bind to the ZEB1 promoter to activate its expression [34]. Insulin-like growth factor binding protein 2 (IGFBP2), another protein from the insulin family, mediates the nuclear localization of p65 in HCC cells. p65 is then able to activate nuclear factor kappa B (NFκB) to promote ZEB1 transcription [35]. Hematological and neurological expressed 1-like (HN1L) acts in concert with Activator Protein-2 gamma (AP-2γ) to promote the transcription of methyltransferase 13, (METTL13), which then cooperates with MYC to transcriptionally upregulate the expression of ZEB1 in HCC cells [36]. Also in HCC cells, Protein phosphatase 1G (PPM1G) alters the alternative splicing of transducin beta like 1 X-linked (TBL1X) favoring the TBL1X-S isoform, which acts as an oncogene by promoting ZEB1 expression [37]. Other EMT-TF, such as Snail, can regulate ZEB1 expression. Indeed, exposure of HCC cells to TPA (O-tetradecanoyl-phorbol 13-acetate) promotes Snail binding to the ZEB1 promoter in a complex involving early growth response 1 (EGR1) and Specificity protein 1 (SP1) [38]. On the contrary, F-box and WD repeat domain containing 8 (FBXW8) acts as a negative regulator of ZEB1 expression by promoting the proteasomal degradation of Palmitoyl-Protein Thioesterase 1 (PPT1), which is an activator of ZEB1 expression in HCC cells [39]. Another F-box protein, F-Box and WD Repeat Domain Containing 7 (FBXW7) negatively regulates ZEB1 expression by inhibiting the mechanistic target of rapamycin kinase (mTOR) signaling in CCA cells [40].
Most of these regulatory mechanisms involve autocrine loops that activate signaling cascades in HCC or CCA cells, but signals coming from the TME can also regulate ZEB1 expression (Figure 2C). In this regard, C-C motif chemokine ligand 5 (CCL5) regulates ZEB1 in liver cancer. In HCC, cancer-associated fibroblasts (CAF) produce CCL5 that, upon binding to tumor cells, activates signals that interfere with the ubiquitination and degradation by the proteasome of hypoxia-inducible factor 1 alpha (HIF1α). Then, HIF1α translocates to the nucleus and binds to the ZEB1 promoter to increase its expression [41]. In CCA, mesenchymal stem cells (MSC) are also able to produce CCL5 upon exposure to pro-inflammatory signals. CCL5 then activates the expression of ZEB1 through a mechanism involving AKT-NFκB signaling activation.
On top of these signaling cascades, different environmental cues have been shown to regulate the expression of ZEB1. Indeed, some etiologic factors that lead to the development of HCC, such as alcohol, hepatitis virus (HCV and HBV) or high-fat diet consumption, can induce ZEB1 expression [42,43,44,45]. In the case of ethanol, the mechanism has been associated with the induction of long non-coding (lnc) RNA lnc171, which in turn sponges microRNA (miRNA or miR) mir-873-5p, a negative regulator of ZEB1 mRNA expression [43]. Ethanol and hepatitis virus have been linked to ZEB1 through a mechanism involving chromosome 8q24 amplification, which leads to MYC expression, that in turn promotes ZEB1 expression [42]. In the case of a high-fat diet, prolonged exposure to fatty acids increased the levels of TGF-β and β-catenin, important EMT inducers and ZEB1 regulators [44]. Nevertheless, other environmental factors besides etiologic factors can regulate ZEB1. Di(2-ethylhexyl) phthalate (DEHP), a worldwide common plasticizer, is often observed in patients because of the continual usage of plastic medical devices. Interestingly, long-term DEHP exposure induces EMT by increasing the expression of EMT-TF, including ZEB1 [46]. High expression of ZEB1 in response to these environmental signals increases the invasiveness, stemness and even resistance to sorafenib, of HCC cells [43,44,45,46].
3.2. Post-Transcriptional Regulation
Besides the above-described regulatory signals, ZEB1 is also under tight post-transcriptional regulations at different levels, including mechanisms that regulate the mRNA export to the cytoplasm, the addition of mRNA modifications, and regulation by non-coding RNAs (ncRNAs), which are RNAs that do not encode proteins but play an important role in post-transcriptional control of gene expression. These ncRNA are classified based on length and structure into lncRNA, miRNA and circular RNA (circRNA). All these post-transcriptional mechanisms are summarized in Figure 3. In terms of RNA modifications, N6-methyladenosine (m^6^A) is the only mRNA modification identified so far in the regulation of ZEB1 in HCC. In this sense, the MYC-associated zinc finger protein (MAZ) is involved in m^6^A methylation in HCC by targeting the transcriptional regulation of key m^6^A enzymes [47]. Indeed, MAZ can upregulate the expression of the methylase Methyltransferase-like protein 3 (METTL3) by directly binding to its promoter. In turn, METTL3 can methylate its targets, among which ZEB1 has been identified in HCC. METTL3-mediated m^6^A methylation of ZEB1 mRNA increases its stabilization [47,48]. Besides writer methylases, the reader YTH domain-containing family protein 3 (YTHDF3) also regulates the stability of ZEB1 in HCC through the m^6^A modification [49]. Another layer of regulation of this process comes from circ_KIAA1429, which is overexpressed in HCC and facilitates the function of YTHDF3 on ZEB1 mRNA [49]. Thus, mechanisms favoring m^6^A methylation of ZEB1 mRNA play a role in increasing the invasion and metastasis of HCC by inducing EMT.
Regarding ncRNAs, the most widely described mechanism is the double-negative feedback loop that ZEB1 forms with the miR-200 family, wherein ZEB1 represses the transcription of miR-200 members while being targeted for degradation by the same miRNAs: a mechanism that allows fine-tuned and dynamic regulation of EMT [50]. However, multiple other miRNAs have been described to regulate ZEB1 in recent years. In addition, there are other upstream signals that may regulate the expression of miRNAs, such as lncRNA, which have gained a lot of interest, as they may regulate miRNAs by acting as sponges and, thus, reducing their availability to target ZEB1. The ZEB1 regulatory mechanisms involving non-coding RNAs are summarized in Table 1.
Apart from these mechanisms, ZEB1 can be regulated post-transcriptionally by ncRNAs without its mRNA levels being affected. A non-POU domain containing octamer binding (NONO) is an RNA-binding protein that facilitates ZEB1 mRNA nuclear export. The lncRNA DIO3OS is generally downregulated in HCC patients. This lncRNA represses ZEB1 by interacting with the NONO protein, thereby restricting NONO-mediated nuclear export of ZEB1 mRNA. Thus, reduced DIO3OS leads to increased ZEB1 protein expression, stemness and HCC progression [51]. Conversely, hsa_circ_0007132 has been found to be increased in patients following lenvatinib treatment and HCC lenvatinib-resistant cell lines. Mechanistically, hsa_circ_0007132 binds to NONO and impairs its ubiquitination-mediated degradation, thereby enhancing the NONO-mediated nuclear export of ZEB1 mRNA. Ultimately, ZEB1 increased protein levels and participates in lenvatinib resistance [52].
It is worth mentioning that while we have knowledge about various types of ncRNAs modulating ZEB1 expression in liver cancer for more than 10 years [53,54], and even longer in other tumors, reports on other posttranscriptional regulatory mechanisms such as RNA modifications (i.e., m^6^A) are as recent as 2025 [47], underscoring the novelty of a growing field in the past few years that will surely help to understand ZEB1’s role, not only in liver cancer but also many other pathological settings.
3.3. Post-Translational Regulation
ZEB1 protein can also undergo post-translational modifications, such as phosphorylation, ubiquitination and acetylation, which influence its stability, localization and transcriptional activity [55]. Very recently, a new form of posttranslational modification has been identified for ZEB1 in HCC, the O-GlcNAcylation. ZEB1 can be O-GlcNAcylated at ser670 by O-GlcNAc transferase (OGT), which increases the protein stability of ZEB1, while O-GlcNAcase (OGA) is able to remove the O-linked N-acetylglucosamine modification. Thus, the expression and activity of OGT and OGA may determine the protein stability of ZEB1 [56]. Nevertheless, in terms of the regulation of ZEB1 expression in liver cancer, most studies are related to ZEB1 degradation by ubiquitination mechanisms (Figure 3). Related to proteasome degradation of ZEB1, deubiquitinase ubiquitin specific peptidase 39 (USP39) and E3 ligase tripartite motif containing 26 (TRIM26) function in an antagonistic pattern in HCC, controlling ZEB1 stability to determine HCC progression. Thus, HCC with a high expression of USP39 show increased ZEB1 expression that determines higher proliferation and migration of HCC cells [57]. Similarly, ubiquitin-specific peptidase 22 (USP22) is involved in the maintenance of ZEB1 stability via its deubiquitinase activity [58]. Another factor involved in ZEB1 degradation by the proteasome is the CRL4-DCAF15 (DDB1 and CUL4 associated factor 15) E3 ubiquitin ligase complex, which recognizes the N-terminal zinc finger domain of ZEB1 to trigger its ubiquitination and posterior degradation, reducing ZEB1 expression and the proliferative and invasive abilities of HCC cells [59]. The ubiquitin-like plant homeodomain (PHD) and really interesting new gene (RING) finger domain-containing protein 1 (UHRF1) protein-associated transcript (UPAT) are also able to promote ZEB1 degradation through the ubiquitin-proteasome pathway in HCC [60]. Besides the proteasome-related mechanisms, ZEB1 protein expression can be reduced by autophagic degradation. In HCC, high mobility group box 1 (HMGB1) promotes homeodomain interacting protein kinase 2 (HIPK2) degradation. HIPK2 is a kinase that plays multifaceted roles in various cellular processes. Among them, HIPK2 regulates ZEB1’s autophagic degradation. Thus, HIPK2 degradation results in protection of ZEB1 from autophagic degradation and increases HCC cell growth. Thus, HMGB1 inhibition can suppress HCC progression via HIPK2-mediated autophagic degradation of ZEB1 [61].
In addition to the above-described regulatory networks controlling ZEB1, the expression of other proteins has been linked to modified expression of ZEB1 without giving precise specifications of the regulatory mechanism. For example, increased expression of HMGB2, 14-3-3ε, POU class 2 homeobox 1 (POU2F1), fibrinogen gamma chain (FGG), RNA-binding motif, Y chromosome (RBMY), stage-specific embryonic antigen 3 (SSEA3), chromobox 6 (CBX6) has been shown to increase EMT, invasiveness and stemness of HCC cells through increasing ZEB1 expression [94,95,96,97,98,99,100], while for F-actin-capping protein subunit alpha-1 (CAPZA1), transient receptor potential cation channel subfamily V member 1 (TRPV1) and liver kinase B1 (LKB1), high expression correlates with low ZEB1 expression and reduced aggressivity of HCC cells [101,102,103]. Similarly, for adrenomedullin (ADM), cullin 4A (CUL4A) and epidermal growth factor (EGF), high expression correlates with high expression of ZEB1 and induced EMT and metastasis in CCA [104,105,106].
4. Role of ZEB1 in Hepatocellular Carcinoma (HCC)
Clinical evaluation showed that ZEB1 is highly expressed in malignant HCC cells compared to normal hepatocytes in 23–65.4% of the patients (Table 2) [91,107,108,109,110]. As an EMT-TF, ZEB1 expression correlated with reduced E-cadherin expression [13,107] and increased Vimentin expression [109]. Furthermore, ZEB1 expression was associated with aggressive clinical parameters, including poorly differentiated tumors, advanced TNM stage, increased number of tumors, multifocal metastases, vascular invasion (Table 3). All of these factors contributed to reduced progression-free survival and overall survival in patients with high ZEB1 expression (Table 2) [91,107,108,109,110].
Preclinical in vitro studies showed that ZEB1 can promote the proliferation, colony formation, sphere formation and migration of HCC cells and markedly enhance tumor progression in vivo in diethylnitrosamine (DEN)-induced models and orthotopic xenografts of human cell lines [91,109,110,112,113]. Mechanistically, multiple factors can be regulated by ZEB1 to lead to these aggressive features, which are indicated in Figure 4. As in patient samples, in HCC cell lines, ZEB1 is able to downregulate the expression of E-cadherin and increase the expression of mesenchymal markers such as Vimentin, matrix metallopeptidase 2 (MMP2) and MMP9, contributing to the migration and invasion of these cells [91,109,110]. ZEB1 is also able to activate the Wnt/β-catenin signaling pathway by upregulating the protein expression levels of β-catenin and its downstream targets c-Myc and Cyclin D1 [91]. Interestingly, ZEB1 has been shown to interact with Yes-associated protein (YAP) to become a transcriptional activator in some aggressive cancers [114]. However, in HCC cells, ZEB1 is able to directly increase the expression of YAP and activate YAP downstream signaling with the upregulation of genes like AXL receptor tyrosine kinase (AXL), connective tissue growth factor (CTGF) and serum deprivation response (SDPR), which contribute to the malignant phenotype [102]. Another transcriptional target of ZEB1 is DEAD-Box 56 (DDX56). Once ZEB1 increases the expression of DDX56, it can exert its downstream actions by upregulating the expression of muscle, intestine and stomach expression 1 (MIST1), which in turn negatively regulates Phosphatase and Tensin Homolog (PTEN) to activate AKT signaling, leading to HCC cell proliferation [115]. Besides classical targets, ZEB1 can control the transcription of ncRNAs. For example, ZEB1 activates the expression of the hsa-microRNA-99b/let-7e/microRNA-125a cluster in the HCC cells to promote invasion and advance liver cancer progression [116].
ZEB1 can also influence the metabolism of HCC cells by upregulating the expression of the muscle isoform of PFKM (phosphofructokinase), a rate-limiting enzyme in glycolysis, which stimulates glycolysis and leads to increased malignant phenotypes through promotion of the Warburg effect [113], a well-known mechanism used by cells undergoing EMT to fuel metabolic needs [117,118]. Similarly, ZEB1 activates the transcription of phosphoglycerate dehydrogenase (PHGDH), which in turn augments the serine synthesis pathway to render the cells more proliferative and invasive and resistant to sorafenib [112]. Activation of the serine biosynthetic pathway is involved in the induction of stemness and chemoresistance across tumor types [119,120]. In HCC cells, PHGDH promotes the translation of mitochondrial DNA-encoded proteins and sustains respiration [121], which helps with meeting the metabolic needs of these cells. This metabolic rewiring has been successfully targeted in other cancers but remains unexplored in HCC [119,120]. Curiously, both PFKM and PHGDH are regulated through non-classic ZEB1 binding sites within their promoter regions [112,113].
Regarding chemoresistance, ZEB1 expression has been shown to increase in HCC cells resistant to oxaliplatin, doxorubicin, sorafenib and bortezomib. These cells are usually enriched in the CD44high/CD24low stem cell population and show augmented expression of Protein kinase C alpha (PKCA) [122]. ZEB1-induced EMT has been linked to resistance to sorafenib [21,46,112], lenvatinib [52] and bortezomib [123], but the exact ZEB1-dependent downstream mechanism has not yet been delineated in these studies.
ZEB1 has also been implicated in the interactions between HCC cells and the TME. ZEB1 interacts with USP22 to coactivate transcriptionally vascular endothelial growth factor A (VEGFA). Then, VEGFA is able to act in a paracrine way to induce cell proliferation, migration and vascular mimicry formation in HCC cells and angiogenesis by acting on endothelial cells [58]. ZEB1 is not only expressed in HCC cells, but also in the TME. Indeed, miR-200b-3p can be encapsulated in exosomes that are taken by macrophages from the TME. In these cells, miR-200b-3p reduces ZEB1 expression, which negatively regulates interleukin 4 (IL4). Thus, increased IL4 levels promote a shift to M2 macrophage phenotype, with increased Pim-1 proto-oncogene, serine/threonine kinase (PIM1) and VEGFA production by the macrophages that act on HCC cells to promote proliferation and metastasis [124].
5. Role of ZEB1 in Cholangiocarcinoma (CCA)
Regarding clinical analysis, nuclear ZEB1 expression has been shown to be highly expressed in malignant intrahepatic CCA cells compared to normal non-neoplastic cells in 20–46.1% of the cases (Table 2) [14,111]. According to its role in EMT, ZEB1 expression showed a significant inverse correlation with E-cadherin expression and a positive correlation with vimentin expression [111]. Interestingly, ZEB1 expression has been associated with programmed cell death ligand 1 (PD-L1) expression in CCA [125]. Although this association has been demonstrated in other tumors [126], there is no mechanistic validation yet in CCA. Importantly, ZEB1 expression was associated with aggressive tumor characteristics, including advanced tumor stage, undifferentiated-type histology, lymph node metastasis and vascular invasion (Table 3) [14,111,127]. Furthermore, patients with high ZEB1 expression showed significantly reduced overall survival rates (Table 2) [111,127].
In vitro, ZEB1 has been shown to promote the proliferation, colony formation, sphere formation and migration of CCA cells and markedly enhance tumor progression in vivo [14,127]. ZEB1 can promote these features by several mechanisms, summarized in Figure 5. On one side, ZEB1 promotes cell migration through the activation of EMT, which leads to downregulation of epithelial markers, such as E-cadherin, and upregulation of mesenchymal markers, such as vimentin [14]. Among the molecular mechanisms related to ZEB1-induced EMT, the feedback loop with miR-200 has been identified in CCA cells [128]. On the other side, ZEB1 increases the stemness of CCA cells by altering the balance of the stem cell markers [14]. ZEB1 upregulates the expression of CD44 [14] (and its alternative splicing) [127], while it downregulates CD24 expression [14], generating a population of cells with high stemness [129,130]. Accordingly, subcutaneous implantation of CCA cells with ZEB1 overexpression produced bigger tumors in nude mice [14]. Curiously, tumors formed by ZEB1 overexpressing cells showed a more abundant stroma than their counterparts. The underlying mechanism involved the direct regulation by ZEB1 of CTGF in CCA cells, which in turn activated CAF proliferation, which could be impaired by a neutralizing antibody [14]. Interestingly, ZEB1 expression has also been detected in CAF from CCA and in different hepatic stellate cell lines. Downregulation of ZEB1 in these cells produced a downregulation of typical activation markers, such as Actin alpha 2, smooth muscle (ACTA2) and Collagen 4A1 (COL4A1). ZEB1 expression in the fibroblasts also impacted CCA cell growth [14]. Indeed, conditioned media from fibroblasts with high ZEB1 expression induced CCA cell growth and activation of the intracellular signaling pathways, including the signal transducer and activator of transcription 3 (STAT3), AKT and ERK. Further analysis showed that ZEB1 upregulated the expression of hepatocyte growth factor (HGF) in liver fibroblasts that induced proliferation of both CCA cells and fibroblasts [14]. These data underscore the role of ZEB1 in regulating tumor cell-CAF crosstalk as promoting tumor dedifferentiation and CAF activation, resulting in tumor progression.
6. Future Perspectives and Therapeutic Opportunities
Liver cancer is one of the leading causes of cancer-related mortality [3], mainly due to the fact that at diagnosis, most patients already present advanced stages of the disease and distant metastasis [1,2]. Consequently, understanding the process of EMT, a key driver of the metastatic cascade, is essential to tackle this important issue. In this context, an increasing number of publications is showing that ZEB1 expression is increased in both HCC and CCA, and it promotes the aggressiveness of these tumors by regulating multiple biological processes related to cell plasticity, including not only EMT, but stemness, therapy resistance and several aspects of the TME development. Thus, understanding the complex regulatory network supervised by ZEB1 may have important clinical and therapeutic implications.
The main issue with exploiting ZEB1’s therapeutic potential is that ZEB1-specific inhibitors are still to be developed. Nevertheless, some indirect approximations have been taken in liver cancer preclinical studies to inhibit ZEB1-related functions (Table 4).
6.1. Chemical Compounds
Farrerol, a bioactive constituent of Rhododendron, is able to suppress TGF-β-mediated migration and invasiveness in HCC cells by inhibiting SMAD2/3 phosphorylation (Figure 2). This impaired phosphorylation led to downregulation of different EMT regulators, including ZEB1 [131]. Similarly, cinobufotalin, a bufadienolide isolated from toad venom, downregulates β-catenin-dependent EMT, which is in part mediated by ZEB1 (Figure 2) [132]. Another report shows that salinomycin, an antibacterial and coccidiostat ionophore, activates FOXO3a, which acts as a negative regulator of the β-catenin, with similar results, reducing ZEB1 expression and EMT in HCC cells (Figure 2). Interestingly, this mechanism mediated by salinomycin is able to revert resistance to doxorubicin in these cells [28]. In CCA, dihydromyricetin (DMY), the main bioactive compound isolated from Ampelopsis grossedentata, significantly reduces proliferation and EMT in tumor cells, through a mechanism involving miR-455-3p upregulation, which in turn targets and inhibits ZEB1 expression [89]. Also in CCA, maraviroc, a CCR5 inhibitor, has been used to impair CCL5-regulated functions upstream of ZEB1 [133]. Additionally, post-translational modifications such as O-GlcNAcylation can be exploited to impair ZEB1 functions. Artesunate, a key antimalarial drug, which originated from the traditional Chinese medicinal plant Artemisia annua, promotes OGA function, which is in charge of removing the O-linked N-acetylglucosamine modification at ser 670, reducing ZEB1 protein stability (Figure 3) [56]. Besides targeting ZEB1 expression, PKC inhibitors have been used to inhibit ZEB1-regulated downstream functions that are mediated by PKCA in HCC cells [122]. The main problem of these approaches is that they do not specifically target ZEB1.
6.2. Neutralizing Antibodies
Neutralizing antibodies against soluble signaling molecules is another regulatory mechanism that can be exploited against ZEB1. For example, neutralizing antibodies against CCL5, produced by MSC or CAF, have been used to inhibit CCL5-regulated processes, including ZEB1 expression [133].
6.3. miRNAs
The potential of miRNAs has been assayed in HCC in a study where miR-708 was used therapeutically to target and downregulate ZEB1 expression [91]. The most accurate therapeutic option available is probably miRNA-based therapy targeting ZEB1 expression. Nevertheless, it is necessary to ensure the specificity of the miRNAs for ZEB1 and to further develop the delivery methods and the stability of the therapeutic agents.
7. Conclusions
In conclusion, ZEB1 is a key regulator of transcriptional plasticity in liver cancer, allowing the cells to adapt to challenges from environmental and therapeutic pressures through the activation of EMT, stemness and chemoresistance programs, which contribute to the aggressivity of the tumors and poorer prognosis of liver cancer patients. Future research should aim to untangle the complexity of the ZEB1-regulated network to identify potential new therapeutic targets that could be integrated with current therapies to improve the evolving treatment landscape of liver cancer patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yang C. Zhang H. Zhang L. Zhu A.X. Bernards R. Qin W. Wang C. Evolving therapeutic landscape of advanced hepatocellular carcinoma Nat. Rev. Gastroenterol. Hepatol.20232020322210.1038/s 41575-022-00704-936369487 · doi ↗ · pubmed ↗
- 2Banales J.M. Cardinale V. Carpino G. Marzioni M. Andersen J.B. Invernizzi P. Lind G.E. Folseraas T. Forbes S.J. Fouassier L. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA)Nat. Rev. Gastroenterol. Hepatol.20161326128010.1038/nrgastro.2016.5127095655 · doi ↗ · pubmed ↗
- 3Bray F. Laversanne M. Sung H. Ferlay J. Siegel R.L. Soerjomataram I. Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J. Clin.20247422926310.3322/caac.2183438572751 · doi ↗ · pubmed ↗
- 4Huang Y. Hong W. Wei X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis J. Hematol. Oncol.20221512910.1186/s 13045-022-01347-836076302 PMC 9461252 · doi ↗ · pubmed ↗
- 5Vaquero J. Guedj N. Claperon A. Nguyen Ho-Bouldoires T.H. Paradis V. Fouassier L. Epithelial-mesenchymal transition in cholangiocarcinoma: From clinical evidence to regulatory networks J. Hepatol.20176642444110.1016/j.jhep.2016.09.01027686679 · doi ↗ · pubmed ↗
- 6Derynck R. Weinberg R.A. EMT and Cancer: More Than Meets the Eye Dev. Cell 20194931331610.1016/j.devcel.2019.04.02631063750 PMC 7672963 · doi ↗ · pubmed ↗
- 7Roy S. Sunkara R.R. Parmar M.Y. Shaikh S. Waghmare S.K. EMT imparts cancer stemness and plasticity: New perspectives and therapeutic potential Front. Biosci. (Landmark Ed.)20212623826510.2741/489333049669 · doi ↗ · pubmed ↗
- 8Tanabe S. Quader S. Cabral H. Ono R. Interplay of EMT and CSC in Cancer and the Potential Therapeutic Strategies Front. Pharmacol.20201190410.3389/fphar.2020.0090432625096 PMC 7311659 · doi ↗ · pubmed ↗