A CRISPR/Cas9-induced restoration of bioluminescence reporter system for single-cell gene expression analysis in plants
Ryohei Ueno, Shogo Ito, Tokitaka Oyama

TL;DR
Researchers developed a CRISPR-based system to restore bioluminescence in plants, enabling detailed single-cell gene expression analysis.
Contribution
A novel CRISPR/Cas9-based system (CiRBS) was developed to restore bioluminescence in transgenic Arabidopsis for single-cell gene expression analysis.
Findings
CiRBS successfully restored bioluminescence in 7.2% of CRISPR/Cas9-transfected cells.
Recombination events via indels were mostly complete within 24 hours of CRISPR/Cas9 induction.
94% of bioluminescence-restored cells had only one chromosome with optimal recombination.
Abstract
Bioluminescence monitoring techniques have greatly contributed to revealing a variety of biological regulatory systems in living organisms, including circadian clocks. In plant science, these techniques are applied to long-term quantitative analyses of gene expression behavior. Transient transfection with a luciferase reporter using the particle bombardment method has been used for bioluminescence observations at the single-cell level. This allows for capturing heterogeneity and temporal fluctuations in cellular gene expression, although bioluminescence could fluctuate according to variation in physiological factors associated with the luciferase reaction. We developed a novel CRISPR/Cas9-induced restoration of bioluminescence reporter system, CiRBS, to monitor cellular bioluminescence from a reporter gene in the genome of transgenic Arabidopsis. In this method, the enzymatic activity…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
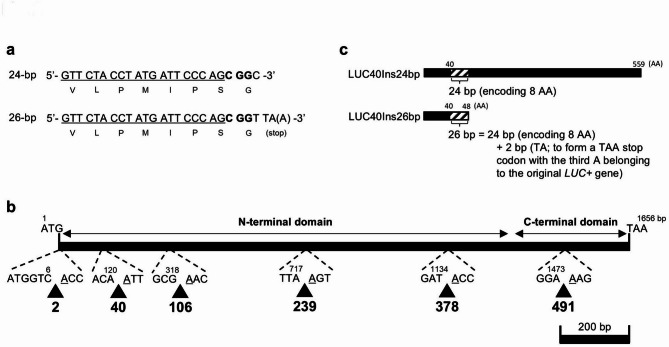 Figure 1
Figure 1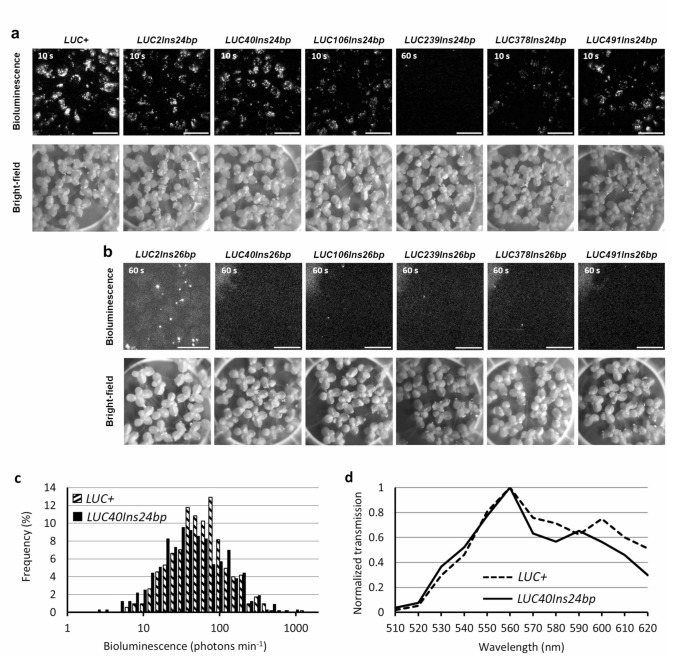 Figure 2
Figure 2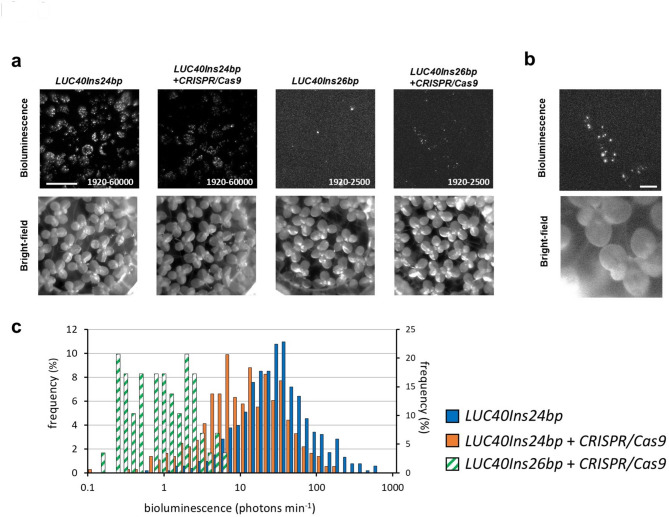 Figure 3
Figure 3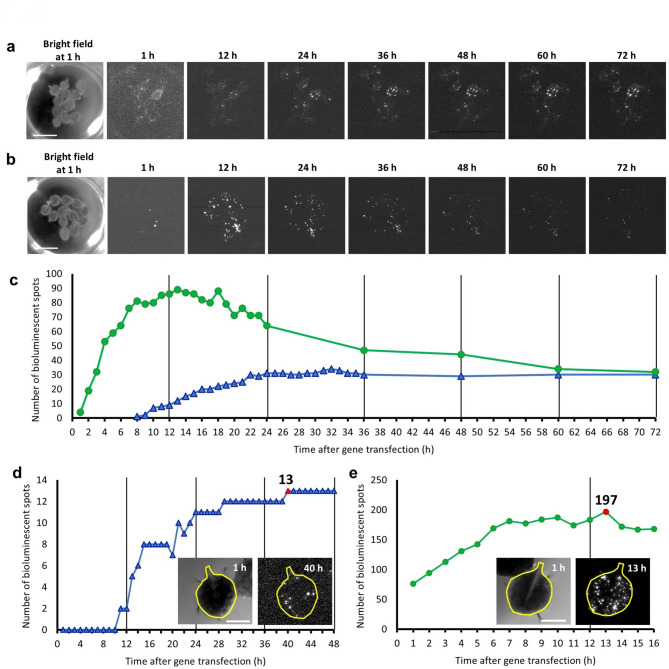 Figure 4
Figure 4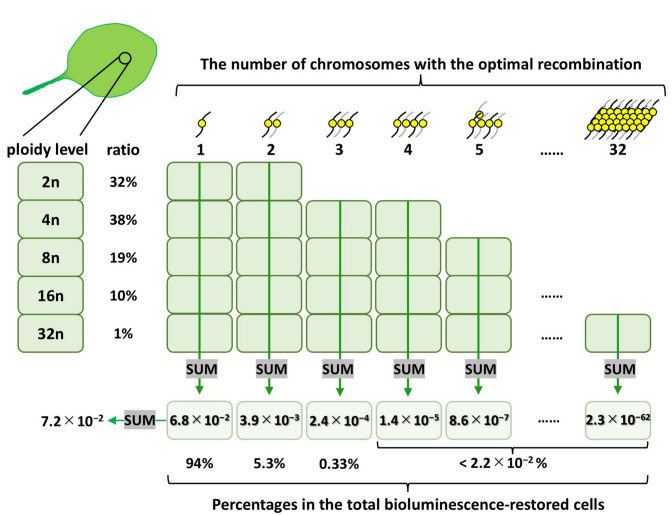 Figure 5
Figure 5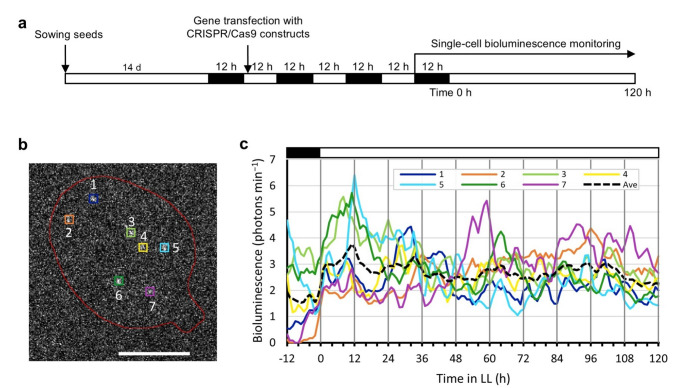 Figure 6
Figure 6- —https://doi.org/10.13039/501100002241Japan Science and Technology Agency
- —https://doi.org/10.13039/501100001691Japan Society for the Promotion of Science
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
Topicsbioluminescence and chemiluminescence research · Light effects on plants · CRISPR and Genetic Engineering
Introduction
Cellular gene expression is the basis of biological systems in multicellular organisms. Gene expression is coordinately regulated but fluctuates stochastically in each cell, and it has been suggested that cellular functions are subject to such noise^1,2^. However, few studies have been performed on the effects of the stochasticity of individual cells on function at the tissue or organism level. The spatial and temporal analysis of gene expression at the single-cell level within the same organ contributes to the understanding of the role of stochastic fluctuation and cell-to-cell heterogeneity in multicellular organisms.
The circadian clock system of plants is a cell-autonomous system^3–5^ composed of cellular oscillators that function via transcription-translation feedback loops of various clock genes^6,7^. Single-cell analyses have revealed cell type-specific circadian behaviors, tissue-specific synchronization characteristics, and coordination among cellular circadian rhythms in an organ^8–10^. In these studies, the accumulation of the product of a clock gene fused with a fluorescent protein gene was monitored in transgenic Arabidopsis plants using confocal microscopy. Given that this reporter system requires an excitation light for fluorescence observation and measurement, it cannot be used to monitor circadian rhythms in darkness. Furthermore, this system is unsuitable for long-term observations with a high temporal resolution of a living plant because of photodamage or unexpected cellular light responses to excitation light^11,12^.
In plants, firefly luciferase (LUC) has been used as a bioluminescent reporter to examine circadian rhythms^13,14^. In contrast to fluorescent reporters, LUC is an enzyme that emits bioluminescence in cells when a luciferin substrate is added^15^, and it is a noninvasive and suitable reporter for the long-term monitoring of gene expression. The bioluminescence rhythms of individual plants, organs, and tissues have been discussed based on the results of transgenic plants expressing LUC under the control of circadian promoters, using either a photomultiplier tube or a high-sensitivity camera^16–20^. Monitoring bioluminescent circadian rhythms at the single-cell level in plants has also been reported. Protoplasts of transgenic Arabidopsis carrying a CIRCADIAN CLOCK ASSOCIATED 1 promoter::luciferase (CCA1::LUC) reporter were observed for their bioluminescence rhythms in culture medium^21^. Furthermore, the cellular circadian behavior in intact plants has been studied using bioluminescence imaging at the single-cell level^22–25^.
Using the particle bombardment method, a bioluminescent reporter can be delivered into plant cells with DNA-coated gold particles^26,27^. Notably, duckweed plants are suitable for long-term bioluminescence monitoring of cells transfected with a reporter using this method. Quantitative single-cell bioluminescence imaging analyses of duckweed plants have demonstrated heterogeneity in clock gene expression among cells within the same organ^22–24,26^. The particle bombardment method also allows co-transfection of a bioluminescent reporter and effector constructs for overexpression, RNAi, or CRISPR/Cas9-induced genome editing^23,28–30^. Although this method is suitable for observing circadian behavior in individual cells, comparative analyses of gene expression between cells are limited. The copy number of the reporter gene introduced into the nucleus is heterogeneous among cells due to heterogeneity in the distance between the nuclei and the introduced gold particles^31^ and possibly due to heterogeneity in the number of reporter plasmids coating the gold particles. Such heterogeneity results in differences in bioluminescence intensities among transfected cells within the 1000-fold range^26^. Moreover, the bioluminescence intensities of cells transiently transfected with a bioluminescence reporter tend to decrease gradually^22^. These problems can be solved by single-cell analysis of the clock gene expression behavior of a LUC reporter gene uniformly introduced into the genome. However, the spatial resolution of the bioluminescence of a stable transgenic plant carrying a LUC reporter under the control of a clock gene promoter is insufficient for the single-cell imaging^18,32^. Therefore, we used a strategy in which cells in a transgenic plant carrying an inactive LUC reporter were subjected to genome editing by introducing a LUC-reactivating effector via particle bombardment to restore bioluminescence at the single-cell level.
LUC proteins, from various luminous insects, consist of N- and C-terminal domains with a flexible linker sequence between the two^15^. The amino acid residues of the active center to which the luciferin substrate and Mg-ATP bind are highly conserved, and mutated LUCs with high enzymatic activity or different colors have been reported^33–36^. These mutated LUCs have been widely used in various bioluminescence imaging experiments^25,28,37^. Hence, we hypothesized that we could either mutate LUC sequences or insert a few amino acid residues in the LUC protein while keeping the luciferase activity, and use it as the bioluminescence reporter in our reactivating strategy.
We chose the CRISPR/Cas9 system, which consists of clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9), as our editing tool^38,39^. A single guide RNA (sgRNA) recognizes and binds to 20-base target sequences, and attracts Cas9, an endonuclease, resulting in double-strand break (DSB)^40^. DSB induces error-prone DNA repair via nonhomologous end-joining (NHEJ) or DNA repair via homology-directed repair (HDR). When repeated sequences exist around the target sequences, HDR occurs immediately after DNA replication and before cell division, and uses homologous double-stranded DNA as template^41^. NHEJ induces point mutations or insertions/deletions (indels) around the DSB site, which alter the functions of the target gene by changing the amino acid sequence, including the introduction of a premature termination codon^42^. A quantitative study on Arabidopsis showed that NHEJ-induced mutations were predominantly 1-bp insertions and short deletions^43^. Gene editing by NHEJ is available for various plant species including Arabidopsis as a high-efficiency mutagenesis technology^44^.
In this study, we developed a CRISPR/Cas9-induced restoration of bioluminescence reporter system (CiRBS) for single-cell expression analysis that enables cell-to-cell comparison of the gene expression behavior of a luciferase reporter in the genome. Transgenic Arabidopsis plants carrying an inactive LUC bioluminescence reporter gene were produced, and cellular bioluminescence was restored by particle bombardment transfection with CRISPR/Cas9 effector constructs to reactivate the LUC gene.
The HDR-based repair of the inactivated mutant reporter (split reporter) gene by the CRISPR/Cas9 system has been used for the analysis of recombination efficiency in plant- and mammalian cells^45,46^. A previous study showed that a modified β-glucuronidase (GUS) reporter (GU-US; carrying a repeated sequence around the target sequence of the sgRNA) expressed in a transgenic potato plant was reactivated through HDR by Agrobacterium-mediated transfection with CRISPR/Cas9-inducible constructs^45^. This reactivation treatment resulted in a 2-fold increase in reporter activity compared to that in the negative controls. In our study, we investigated the conditions under which the modified LUC exhibited null activity, and such activity was restored by indels through NHEJ. We first identified a site in the LUC gene at which a 24-bp sequence insertion, including the sgRNA target, retained the luciferase activity of the gene product. We then constructed a modified LUC with a 26-bp insertion at the same site to inactivate luciferase activity by frameshift and stop codon. This 26-bp insertion also included the sgRNA target sequence. Finally, transgenic Arabidopsis plants carrying this construct were created. The modified LUC was reactivated by transfection with sgRNA- and Cas9-expression constructs (CRISPR/Cas9 constructs) using particle bombardment. Hence, we successfully monitored the bioluminescence of the reporter gene in the genome at the single-cell level.
Results
Design of six insertion sites in the luciferase (LUC) gene for CRISPR/Cas9 target sequences
We generated mutant LUC constructs with specific 24-bp or 26-bp insertions for the CiRBS (Fig. 1a). LUCXAAIns24bp and LUCXAAIns26bp included 24-bp and 26-bp insertions after the X_AA_th codon from the start codon (ATG) in the wildtype LUC gene (LUC+), respectively (Fig. 1b). The first 20-bp sequence was the sgRNA target that was used for the analysis of the efficiency of CRISPR/Cas9 system in potato^45^; no sequence matching this 20-bp sequence was found in the Arabidopsis genome. The same PAM sequence (CGG) followed the target sequences. Thus, the first 23-bp sequence was identical in all insertions. LUCXAAIns26bp insertions resulted in LUC deletion mutants due to the formation of a stop codon and a frameshift at the end of each insertion (Fig. 1c). The third nucleotide (A) of the stop codon (TAA) belonged to the original LUC+ gene. LUCXAAIns26bp insertions were expected to generate null mutants, whose bioluminescence activity could be restored by CRISPR/Cas9-induced indels in the insertion sequences themselves. LUCXAAIns24bp represented revertant LUC mutants. LUCX_AA_Ins24bp proteins included an 8-AA sequence (V-L-P-M-I-P-S-G) at the insertion site. We selected six insertion sites after codons 2nd, 40th, 106th, 239th, 378th, and 491st from the LUC+ start codon.
Fig. 1 Schematic representation of LUCXAAIns24bp and LUCXAAIns26bp inserts. (a) Nucleotide and amino acid (translated below each codon) sequences of the insertion in LUCXAAIns24bp (top) and LUCXAAIns26bp (bottom). The target sequences of the sgRNA of the CRISPR/Cas9 system are underlined. Characters in bold indicate the PAM sequence CGG. The adenine in parentheses in the LUCXAAIns26bp sequence represents the first nucleotide of the original LUC+ gene, which is involved in the generation of a stop codon at the insertion site. (b) Map of insertion sites. Arrowheads indicate the insertion sites for LUCXAAIns24bp and LUCXAAIns26bp. The number below each arrow represents the codon (X_AA_) next to which either LUCXAAIns24bp or LUCXAAIns26bp were inserted. The N- and C-terminal domain coding regions are represented by horizontal lines^15^. (c) Schematic representation of the proteins encoded by LUCXAAIns24bp (top) and LUCXAAIns26bp (bottom) inserted at the 40th codon (LUC40Ins24bp and LUC40Ins26bp, respectively). Shaded boxes represent the 8-AA inserted regions.
Selection of the LUC40Ins26bp for CiRBS
To determine which LUCX_AA_Ins24bp proteins retained the highest bioluminescence activity, we quantified the bioluminescence intensity of each LUCXAAIns24bp variant. We transiently transfected duckweed (Lemna japonica) plant cells with a CaMV35S::LUCXAAIns24bp construct using particle bombardment. CaMV35S::LUCXAAIns24bp contains a LUCXAAIns24bp coding sequence under the control of the Cauliflower mosaic virus 35S (CaMV35S) promoter. Duckweed plants are suitable materials for bioluminescence imaging at the single-cell level because of their tiny flat bodies^22,26,32^. Bioluminescence imaging was performed 24–30 h after gene transfection. Bioluminescent spots were observed in all CaMV35S::LUCXAAIns24bp transfected cells except for those with CaMV35S::LUC239Ins24bp (Fig. 2a). We then checked for the loss of bioluminescence activity in each CaMV35S::LUCXAAIns26bp construct. Almost no bioluminescent spots were observed for any of the CaMV35S::LUCXAAIns26bp-expressing cells except for those with CaMV35S::LUC2Ins26bp (Fig. 2b).
Fig. 2 Bioluminescence of LUCXAAIns24bp and LUCXAAIns26bp. (a, b) Bioluminescence (top) and bright-field (bottom) images of duckweed plant cells transfected with (a) CaMV35S::LUCXAAIns24bp or (b) CaMV35S::LUCXAAIns26bp. The exposure time for capturing bioluminescence is shown in each image. The signal range for the bioluminescence images was fixed to (a) 1920–5000 or (b) 1920–2500. The signal range of raw 16-bit images was 0–65535, whereas that of the background was 1920. Bars: 10 mm. (c) Frequency distribution (%) of cellular bioluminescence intensities for CaMV35S::LUC+ (n = 526) and CaMV35S::LUC40Ins24bp (n = 314). The mean bioluminescence intensity of CaMV35S::LUC40Ins24bp is compared to that of CaMV35S::LUC+ (p = 0.0026 by Welch’s two-tailed t-test). Twenty-five duckweed colonies were observed in one experiment. (d) Bioluminescence spectra of LUC+ and LUC40Ins24bp in duckweed plants. The transmitted bioluminescence intensities were normalized to that at 560 nm and plotted. Bioluminescence was filtered with a series of band-pass filters and quantified^25^.
We quantified the bioluminescence intensities of individual spots for each CaMV35S::LUCXAAIns24bp- and CaMV35S::LUC+-expressing cells (Fig. 2c; Supplementary Fig. S1). At this time, the mean bioluminescence intensity of CaMV35S::LUC40Ins24bp was higher than that of any other CaMV35S::LUCXAAIns24bp constructs, and the bioluminescence intensity distribution in CaMV35S::LUC40Ins24bp and CaMV35S::LUC+ cells was similar (Fig. 2c). Although the mean bioluminescence intensity of CaMV35S::LUC2Ins24bp was also similar to that of CaMV35S::LUC40Ins24bp (Supplementary Fig. S1; p = 0.061, Welch’s two-tailed t-test), CaMV35S::LUC2Ins26bp produced undesirable bioluminescent spots (Fig. 2b). We also examined the emission spectra of CaMV35S::LUCXAAIns24bp and CaMV35S::LUC+ and confirmed that those of LUC40Ins24bp and LUC+ were similar (Fig. 2d; Supplementary Fig. S2). Then we concluded that the insertion at the 40th codon was the best among the six candidate sites, and used LUC40Ins26bp for further CiRBS experiments.
We then examined NHEJ-induced restoration of bioluminescence of LUC40Ins26bp. In order to test the effectiveness of our sgRNA, duckweed cells were transiently co-transfected with both CaMV35S::LUC40Ins24bp and CRISPR/Cas9 constructs by particle bombardment. The mixture of pENTR_AtU6-26::sgRNA-LUCXAA and the plasmid expressing Cas9 (pUC18_PcUBQ4-2::Cas9)^23,47^ is defined as the CRISPR/Cas9 constructs in this study. The cellular bioluminescence intensities of CaMV35S::LUC40Ins24bp in the co-transfected samples were considerably lower than in those carrying CaMV35S::LUC40Ins24bp only (Fig. 3a and c). Furthermore, co-transfection with a nontargeting mixture containing pENTR_AtU6-26::sgRNA and pUC18_PcUBQ4-2::Cas9 (nonsense-CRISPR/Cas9 constructs) did not lead to decreased bioluminescence intensities (Supplementary Fig. S3). We then transiently co-transfected duckweed cells with CaMV35S::LUC40Ins26bp and the CRISPR/Cas9 constructs. The number of bioluminescent spots dramatically increased (Fig. 3a and b), whereas few bioluminescent spots were observed for cells transfected with CaMV35S::LUC40Ins26bp without the CRISPR/Cas9 constructs (Figs. 2b and 3a). Given that NHEJ-induced mutations are predominantly 1-bp insertions and short deletions^43^, luciferase activity restoration was likely triggered by these indels at the insertion site of LUC40Ins26bp. The geometric mean of bioluminescence intensities of restored-LUC40Ins26bp was 3% of that of LUC40Ins24bp (Fig. 3c). This suggests that a small portion of the introduced LUC40Ins26bp copies by particle bombardment was restored through NHEJ. We also transiently co-transfected duckweed cells with both CaMV35S::LUC40Ins26bp and the nonsense-CRISPR/Cas9 constructs, but no bioluminescent spots were observed (Supplementary Fig. S3).
Fig. 3. Restoration of LUC40Ins26bp bioluminescence by co-transfection with CRISPR/Cas9 constructs. (a) Bioluminescence (top) and bright-field (bottom) images of duckweed plants transfected with reporter and CRISPR/Cas9 constructs (indicated above each set of images). Signal ranges are shown in each bioluminescence image. Exposure time: 60 s; Bar: 10 mm. (b) Close-up images of duckweed plants co-transfected with CaMV35S::LUC40Ins26bp and CRISPR/Cas9 constructs. Exposure time: 180 s. The signal range: 1920–2500. Bar: 2 mm. (c) Frequency distribution (%) of cellular bioluminescence intensities for CaMV35S::LUC40Ins24bp (blue bars; n = 529; left side y-axis), CaMV35S::LUC40Ins24bp and CRISPR/Cas9 constructs (orange bars; n = 364; left side y-axis), and CaMV35S::LUC40Ins26bp and CRISPR/Cas9 constructs (green shaded bars; n = 29; right side y-axis). The mean bioluminescence intensity of CaMV35S::LUC40Ins24bp and CRISPR/Cas9 constructs is compared to that of CaMV35S::LUC40Ins24bp (p = 3.7 × 10^− 42^ by Welch’s two-tailed t-test). Twenty-five duckweed colonies were observed in one experiment.
We further tested the bioluminescence restoration of LUC40Ins26bp using a plant-derived promoter commonly employed in physiological experiments. We chose a circadian promoter of the Arabidopsis clock gene, CIRCADIAN CLOCK ASSOCIATED 1 (AtCCA1)^4^. Duckweed plants transfected with AtCCA1::intronLUC+ or AtCCA1::LUC40Ins24bp via particle bombardment showed a bioluminescence circadian rhythm^48^ (Supplementary Fig. S4). In contrast, the bioluminescence of plants transfected with AtCCA1::LUC40Ins26bp did not show a circadian rhythm, and its intensities were close to the background level. Notably, co-transfection with AtCCA1::LUC40Ins26bp and CRISPR/Cas9 constructs restored bioluminescence and exhibited circadian rhythms similar to those of AtCCA1::intronLUC+ and AtCCA1::LUC40Ins24bp, although with lower intensities. Therefore, the LUC40Ins26bp reporter system is likely to function without altering the physiological relevance of the promoter.
Cellular bioluminescence restoration in transgenic Arabidopsis carrying CaMV35S::LUC40Ins26bp
We tested CiRBS in transgenic Arabidopsis plants carrying CaMV35S::LUC40Ins26bp integrated in the genome. We selected a T3 homozygous line, LUC40Ins26bp #28-3, which strongly expressed LUC40Ins26bp from a single locus (Supplementary Fig. S5). We transiently transfected detached leaves of the transgenic line with CRISPR/Cas9 constructs using particle bombardment. Immediately after transfection, the leaves were floated on a luciferin-containing medium and monitored for cellular bioluminescence under constant dark conditions for one week. We successfully monitored restored bioluminescence at the single-cell level (Fig. 4a; Supplementary Video S1). This restored bioluminescence appeared to be stably maintained when compared to that in Col-0 detached leaves with conventional transient transfection with CaMV35S::LUC+ (Fig. 4b; Supplementary Video S1). Few bioluminescent spots were detected when nonsense-CRISPR/Cas9 constructs were used for transfection, and no bioluminescent spots when only pENTR_AtU6-26::sgRNA-LUCXAA or only pUC18_PcUBQ4-2::Cas9 were used (Supplementary Video S2).
Fig. 4. Time-course of the bioluminescence restoration events in transgenic Arabidopsis carrying CaMV35S::LUC40Ins26bp (line #28-3). (a,** b**) Low-magnification bioluminescence images of the (a) bioluminescence-restored CaMV35S::LUC40Ins26bp and (b) transiently-transfected CaMV35S::LUC+ Arabidopsis leaves. Bioluminescence images were hourly captured under constant dark conditions. Time after gene transfection is indicated. Exposure time: (a) 150 and (b) 60 s. The signal range was fixed to 1920–2200. Bar: 10 mm. (c) Number of bioluminescent spots with over 2000 signals in (a) (blue △) and (b) (green 〇). (d,** e**) Number of bioluminescent spots in (d) restored-bioluminescence CaMV35S::LUC40Ins26bp and (e) transiently-transfected CaMV35S::LUC+ Arabidopsis leaves. The bioluminescent spots in the leaf with the highest gene transfection efficiency for each treatment were counted. Red symbol: maximum number of bioluminescent spots. The bright-field (left) and bioluminescence (right) images of the corresponding leaves (together with a representation of the shape of each leaf) are shown. Bioluminescence monitoring conditions were the same as in (a, b) except for the magnification used for a better visualization of the spots. The areas of the leaves are (d) 16.9 and (e) 18.5 mm². Bars: 2.5 mm.
To quantitatively analyze the timing of bioluminescence emergence, we counted the bioluminescent spots in a series of time-lapse images for Fig. 4a and b. These bioluminescent spots were detected immediately after CaMV35S::LUC+ transient transfection of Col-0 detached leaves (Fig. 4b and c). The number of spots increased after 12 h, and then gradually decreased. The transiently transfected reporter appeared to be unstable in cells. In contrast, when detached leaves of the LUC40Ins26bp #28-3 line were transfected with the CRISPR/Cas9 constructs, we first detected a restored bioluminescent spot at 8 h, and the number of spots gradually increased for up to 24 h (Fig. 4a and c). Thereafter, the number was maintained, and bioluminescence appeared to persist. This indicated that most bioluminescence restoration events were completed within 24 h of gene transfection with CRISPR/Cas9, and cellular bioluminescence was stably maintained. In these leaves, it took a longer time for bioluminescent spots to emerge compared to those observed after transient transfection with CaMV35S::LUC+, probably because of the multiple molecular events required for such restoration, including the expression of CRISPR/Cas9 constructs, DSB, NHEJ, and recombinant LUC40Ins26bp expression. Although some bioluminescent spots emerged several days after CRISPR/Cas9 construct transfection (Supplementary Video S1), bioluminescence was basically restored the day after transfection.
We tried to estimate the efficiency of bioluminescence restoration by NHEJ in the transfected cells, and the apparent efficiency was defined as the ratio of the number of restored bioluminescent spots to the number of transiently transfected cells in LUC40Ins26bp #28-3 leaves. However, we were unable to simultaneously detect these two cell types in the same leaf. To estimate the ratio, we counted the maximum number of bioluminescent spots in a leaf of wild-type transfected with CaMV35S::LUC+, and also counted the maximum number of restored bioluminescent spots in a leaf of the LUC40Ins26bp #28-3 line transfected with the CRISPR/Cas9 constructs (Fig. 4d and e; Supplementary Video S3). The number of bioluminescent spots was 197 at 13 h for wild-type leaves (Fig. 4d), and 13 at 40 h for LUC40Ins26bp #28-3 leaves (Fig. 4e). Based on these numbers, we estimated the apparent efficiency as follows:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$\:\frac{\raisebox{1ex}{$13$}\!\left/\:\!\raisebox{-1ex}{$27045$}\right.}{\raisebox{1ex}{$197$}\!\left/\:\!\raisebox{-1ex}{$29568$}\right.}=\frac{4.8\times\:{10}^{-4}}{6.7\times\:{10}^{-3}}=7.2\times\:{10}^{-2}.$$\end{document}The number of bioluminescent spots was corrected for leaf size. Thus, it was presumed that 7.2% of total transfected cells restored the luciferase activity in our experimental conditions.
We next estimated the probability of a recombination event that restored luciferase activity at the LUC40Ins26bp locus in each chromosome. In Arabidopsis leaves, the ploidy levels of cells range from 2C to 32C owing to endoreduplication^49,50^. The reported ratio in epidermal cells is 2n:4n:8n:16n:32n = 0.32:0.38:0.19:0.10:0.01^49^. In this study, we referred to this ratio to calculate the probability of recombination events that restore bioluminescence. Given our estimation that bioluminescence-restored cells accounted for 7.2% of the cells transfected with CRISPR/Cas9 constructs, the optimal recombination rate (α) at the LUC40Ins26bp locus was 1.37% (Supplementary Fig. S6; Supplementary Spreadsheet S1). It was also calculated that 94% of the bioluminescence-restored cells carried only one chromosome with the optimal recombination (Fig. 5; Supplementary Spreadsheet S2). Therefore, the bioluminescence intensities of most cells were presumed to reflect the expression level of the LUC40Ins26bp locus on a single chromosome in the single-cell CiRBS.
Fig. 5 Estimation of the ratios of transfected cells carrying various number ofchromosomes with the optimal recombination for bioluminescence restoration. We referred to the ploidy profile of Arabidopsis leaf epidermal cells (2n:4n:8n:16n:32n = 0.32:0.38:0.19:0.10:0.01) reported by Kawade and Tsukaya, 2017^49^. Based on a Poisson distribution with the probability of the optimal recombination (α = 0.0137; see Supplementary Fig. S6; Supplementary Spreadsheet S1), we calculated the ratios of bioluminescence-restored cells to transfected cells in every combination of chromosome number and ploidy level (Supplementary Spreadsheet S2). The ratios of bioluminescence-restored cells carrying 1n–32n chromosomes with the optimal recombination are represented. The percentages of cells carrying 1, 2, 3, and > 4 chromosomes with the optimal recombination among all bioluminescence-restored cells are also represented.
Observation of cellular bioluminescence traces by CiRBS
To demonstrate the single-cell analysis using CiRBS, we transiently transfected cells into the detached leaves of LUC40Ins26bp #28-3 with CRISPR/Cas9 constructs using particle bombardment, and monitored the restored bioluminescence of LUC40Ins26bp for a week. Leaves were collected from plants grown under 12-h light/12-h dark conditions before gene transfection (Fig. 6a). We quantified the bioluminescence intensities of the seven spots whose bioluminescence continued during the measurements (Fig. 6b and c; Supplementary Video S4). The bioluminescence of each spot appeared to fluctuate asynchronously in the same detached leaf, suggesting that fluctuations in gene expression in the genome of individual cells can be analyzed using CiRBS. The mean bioluminescence intensities of the seven spots remained nearly constant under constant light. In another strain (LUC40Ins26bp #38-2), we observed cellular bioluminescent traces similar to those in Fig. 6c (Supplementary Fig. S7; Supplementary Video S5). Intensities were comparable within a 10-fold range at each time point. Interestingly, transient transfection with CaMV35S::LUC+ resulted in a large variation in bioluminescence intensity, over a 100-fold range, among cells (Fig. 2c). These results suggest that the observation of cellular bioluminescence by CiRBS allows us to compare cellular gene expression behavior, including expression levels and stochastic fluctuations between cells.
Fig. 6. Bioluminescence traces of restored-LUC40Ins26bp at the single-cell level. (a) Time schedule of the monitoring experiments. Gene transfection of cells in detached leaves with CRISPR/Cas9 constructs was conducted by particle bombardment. Two days after gene transfection, the leaf showing the largest number of bioluminescent spots was selected for bioluminescence monitoring. Time 0 indicates the start of the constant light. Hourly bioluminescence imaging was performed during the monitoring. White and black boxes represent light and dark conditions, respectively. (b) Snapshot of bioluminescent spots (colored □) at 3 h. The seven bioluminescent spots of interest were numbered from the distal to the proximal part of the leaf. The shape of the leaf is roughly represented. Bar: 2 mm. (c) Bioluminescence traces of the seven spots. Quantified intensities (3-h moving average) are plotted for each bioluminescent spot shown in (b). Mean intensities for the seven spots are represented by a black dotted line.
Discussion
We successfully developed a CiRBS, which enabled gene expression analysis at the single-cell level, and demonstrated fluctuations in luciferase gene expression driven by the CaMV35S promoter on the genome of individual cells. This CiRBS is based on the NHEJ of the CRISPR/Cas9 system to restore the luciferase activity of a loss-of-function LUC mutant. Previous reports have shown gene restoration through HDR triggered by CRISPR/Cas9 between repeated sequences^45,46^. Therefore, we first attempted this strategy and produced a modified LUC gene with 702-bp repeated sequences before and after the sgRNA target sequence in the coding region (named LU_UC gene; Supplementary Fig. S8). Although LU_UC was expected to be a complete loss-of-function gene, transient transfection of leaves with CaMV35S::LU_UC resulted in a much stronger bioluminescence emission than the background signal of the sample. The retained luciferase activity of LU_UC restricted its usage in the CiRBS. In addition, transient co-transfection with LU_UC and CRISPR/Cas9 constructs did not considerably increase the bioluminescence intensity (Supplementary Fig. 9). Considering that HDR is more frequently induced during cell division^41^, NHEJ is likely to be more dominant than HDR in mature cells, which are the targets of transfection by particle bombardment^51^.
We then aimed to develop a simple reporter gene with little or no bioluminescence background. When comparing the LUC sequences of various luminous insects, the active sites were found to be highly conserved^15^. To construct CiRBS using NHEJ, we investigated the sites at which a short amino acid insertion would not affect luciferase activity. LUCXAAIns24bp construction, which included a 24-bp insertion sequence (itself including the sgRNA target sequence) inserted at certain X_AA_ sites of the LUC+ gene, showed various enzymatic activities depending on the X_AA_ insertion site (Fig. 2a and c; Supplementary Fig. S1). LUC2Ins24bp and LUC40Ins24bp showed bioluminescence intensities comparable to those of wild-type luciferase LUC+. This suggests that luciferase is robust against mutations at these sites. In contrast, LUC239Ins24bp was a mutant LUC protein in which the 24-bp sequence was inserted near the active site, resulting in activity loss. Additionally, it has been reported that the structure around the active site is related to the color of LUC bioluminescence^52^. Therefore, we examined the bioluminescence spectra of LUCX_AA_Ins24bp proteins. While LUC106Ins24bp exhibited a strong red emission (> 590 nm), all other LUCX_AA_Ins24bp did not considerably change their spectra (Supplementary Fig. S2; Fig. 2d).
By introducing both a frameshift and a stop codon (TAA) at the end of the 26-bp insertion, we succeeded in losing the luciferase activity of LUCX_AA_Ins26bp proteins, except for that of LUC2Ins26bp (Fig. 2b). We expected that LUC2Ins26bp would lose its activity because it only encodes a 10-AA peptide sequence from the start codon of the native protein. Its luciferase activity might be caused by an N-terminal 30-AA deleted mutant LUC, starting from an internal in-frame Met codon. It should be noted that LUC491Ins26bp lost its enzymatic activity, although most of the LUC sequence remained intact (Figs. 1 and 2). This finding supports a previous report indicating that the C-terminal domain of luciferase plays a crucial role in the stable binding of luciferin and the subsequent luminescence reaction^52^.
We selected LUC40Ins26bp as the reporter gene for CiRBS because the 24-bp insertion maintained luciferase activity, whereas the 26-bp insertion lost it (Fig. 2a and b). The amplitudes of the bioluminescence rhythms of AtCCA1::intronLUC+ and AtCCA1::LUC40Ins24bp were similar (Supplementary Fig. S4), suggesting that the stability of LUC40Ins24bp was similar to that of LUC+^53^. However, we detected a few bioluminescent spots on a small number of fronds (leaf-like structures) when duckweed plants were transfected with CaMV35S::LUC40Ins26bp (Fig. 3a). We tested the possibility that an alternative translation start site located after the insertion (cLUC40) could be utilized to produce a partially deleted luciferase protein that retained its enzymatic activity when in the presence of the N-terminal polypeptide stopped at the insertion site (nLUC40) (Supplementary Fig. S10). However, co-transfection with two genes encoding these polypeptides did not restore bioluminescence activity. Alternatively, cells with a reversion mutation in the insertion region may have partially regained luciferase activity. However, the luminescence of these bioluminescent spots tended to dampen quickly, suggesting that a stable mutation was unlikely. Transcription errors that induce reversion of luciferase activity may occur with a low probability^54^. Therefore, these transcriptional errors may have caused the low background luminescence signals observed after adding luciferin to the medium (Fig. 4a). Notably, the background and stochastic luminescence rarely affected the performance of single-cell CiRBS monitoring and analysis (Supplementary Video S1; Supplementary Video S2).
Based on our data regarding the emergence of bioluminescence (Fig. 4), we estimated the time required for recombination processes induced by transfection with the CRISPR/Cas9 constructs using particle bombardment. After the cells in Col-0 leaves were transiently transfected with CaMV35S::LUC+, bioluminescent spots were detected within 1 h, suggesting that the expression of genes transfected via particle bombardment occurred rapidly. In contrast, it was previously reported that indel mutations were first observed 6 h after introducing CRISPR/Cas9 into tomato protoplasts and their number increased for up to 2–3 d^55^. Consistent with this report, it took 8 h to detect the first restoration of bioluminescence after gene transfection using our CiRBS. Most bioluminescent spots emerged within 24 h, and the bioluminescence persisted during monitoring (Fig. 4c). This indicated that transient expression of CRISPR/Cas9 constructs was enough to induce restoration of the LUC40Ins26bp reporter for long-term bioluminescence monitoring as stable transgenic cells. Interestingly, a small number of bioluminescent spots appeared several days after gene transfection (Supplementary Video S5). Various events, including DSB, NHEJ, and transcription-translation of recombinant genes, likely contributed to the delay in bioluminescence emergence.
In this study, we successfully detected cellular bioluminescence from a luciferase driven by the constitutive promoter CaMV35S as a model for the single-cell analysis using CiRBS (Fig. 6; Supplementary Fig. S7). Cellular bioluminescence intensity was comparable within a 10-fold range of the mean value. In Arabidopsis leaves, the ploidy levels of cells range from 2C to 32C owing to endoreduplication^49,50^. The variation in cellular bioluminescence intensities might be partly due to differences in the number of restored LUC genes among the cells. However, we estimated that 94% of the bioluminescence-restored cells carried only one chromosome with the optimal recombination (Fig. 5; Supplementary Spreadsheets S2); the bioluminescence intensities of most cells reflected the expression level of a single reporter locus in one chromosome. Consequently, single-cell bioluminescence intensity comparison analyses using CiRBS will be highly reliable.
Single-cell monitoring using a reporter system is a powerful tool for analyzing stochastic gene expression and cell-to-cell heterogeneity within the same tissue^1,2,10,22,23^. We observed that restored bioluminescent spots within the same leaf exhibited fluctuations that were asynchronous between cells (Fig. 6c; Supplementary Fig. S7). These fluctuations might reflect stochastic variation in the transcriptional activity of the constitutive promoter CaMV35S in the genome. However, it should be noted that bioluminescence fluctuations could arise not only from transcriptional dynamics but also from variation in physiological factors associated with the luciferase enzymatic reaction. Such factors include cytoplasmic luciferin, magnesium ions, protons, ATP, and oxygen, all of which can influence luciferase activity^56^. Fluctuations in the production, consumption, or uptake of these factors may therefore affect the bioluminescence traces. Interestingly, circadian fluctuations in some of these factors have been reported in duckweed^57^. By comparing bioluminescence fluctuations observed in CiRBS using various promoters, it should be possible to dissect promoter-specific contributions to the observed variability. Furthermore, a dual-color bioluminescence monitoring system that enables simultaneous detection of two reporters in the same cell could be integrated into CiRBS to distinguish transcriptional dynamics from fluctuations in physiological factors^25,28^.
CiRBS is expected to be applicable to any plant species that can be stably transformed, and it would facilitate single-cell gene expression analyses from various physiological perspectives. Employing circadian-, tissue-specific-, or stimulus-responsive promoters in CiRBS could be particularly valuable, as this would allow fluctuations in the associated physiological processes of individual cells to be observed. When particle bombardment is used for CiRBS, CRISPR/Cas9 constructs are primarily delivered into cells located near the tissue surface^26^. Consequently, this gene delivery method restricts the application of CiRBS to promoters that can be evaluated in those cells. As an alternative to transient gene transfection by particle bombardment, an inducible CRISPR/Cas9 construct that is stably integrated into the genome of plant could be used for CiRBS. For instance, placing the Cas9 gene under the control of a chemically inducible promoter^58^ could allow regulation of the efficiency of bioluminescence restoration.
Observation of cellular bioluminescence by CiRBS allows us to compare cellular gene expression behavior, including expression levels and stochastic fluctuations between cells. In other words, the CiRBS at a single-cell level will make it possible to simultaneously capture “fluctuations in expression (temporal changes within the single-cell)” and “heterogeneity (differences between cells),” which are currently difficult to analyze simultaneously.
Methods
Plant material and growth conditions
Duckweed plants, Lemna japonica 5512 [Lemna minor or Lemna aoukikusa in previous studies: the Rutgers Duckweed Stock Cooperative (http://www.ruduckweed.org/)] were obtained from a small pond in a botanical garden of Hokkaido University (Hokkaido, Japan)^59^. They were maintained in Non-flowering (NF) medium with 1% sucrose at 25 ± 1 °C under constant light conditions as previously described^25^. White light (30–35 µmol m^− 2^·s^− 1^) was supplied by fluorescent lamps (FLR40SEX-W/M/36-HG; NEC, Tokyo, Japan). L. japonica plants were grown on 60–80 mL of NF medium in 300-mL Erlenmeyer flasks plugged with cotton to prevent contamination. New stock cultures were prepared once every two weeks, and well-grown plants were used for the experiments.
Arabidopsis thaliana plants (Col-0 and transgenic CaMV35S::LUC40Ins26bp/Col-0) were grown in plates containing 0.5× Murashige and Skoog (MS) medium with 0.8% agar and 1% sucrose at 22 °C under constant light conditions as previously described^21^. White light (30–35 µmol m^− 2^·s^− 1^) was supplied by fluorescent lamps (FLR40SEX-W/M/36-HG; NEC). We used 14-day-old Arabidopsis thaliana plants and cut the first and second leaves with sterile scissors for particle bombardment. Arabidopsis seeds were sterilized using the following procedure: seeds were collected in sterilized 1.5 mL Eppendorf tubes and washed once with 70% ethanol, the supernatant was removed, a seed sterilization solution (5% bleach [Kao, Tokyo, Japan] and 0.05% Triton x-100 [nacalai tesque, Kyoto, Japan]) was added, and the tubes were shaken for 10 min. The seeds were then washed three times with sterile water, and 1 mL of sterile water was added to the tubes, which were shaded at 4 °C for 2 d before culturing on MS medium. Seed sterilization for the selection of transgenic plants was performed using the chlorine gas method (10 mL HCl to 100 mL 5% bleach)^60^. The seeds were sterilized in a sealed desiccator for 6 h.
Gene transfection constructions
pUC18_CaMV35S::LUCXAAIns24bp and pUC18_CaMV35S::LUCXAAIns26bp were constructed as follows: We first constructed pENTR_LUC+ by inserting the luciferase coding sequence (LUC+; Promega, Madison, WI, USA) into the cloning site of the pENTR/D-TOPO vector (Thermo Fisher Scientific, Waltham, MA, USA). Then, we constructed pENTR_LUCXAAIns24bp/26bp, a derivative of pENTR_LUC+.
pENTR_LUC2Ins24bp/26bp were generated using a KOD-Plus-Mutagenesis Kit (TOYOBO, Osaka, Japan) with a set of specific primers (Supplementary Table S1). Circularization was performed using the In-fusion method (In-Fusion^®^ Snap Assembly Master Mix; Takara Bio, Shiga, Japan). To construct pENTR_LUC40Ins24bp/26bp, pENTR_LUC106Ins24bp/26bp, pENTR_LUC239Ins24bp/26bp, pENTR_LUC378Ins24bp/26bp, and pENTR_LUC491Ins24bp/26bp, we prepared three DNA fragments: 5’-part-LUC, 3’-part-LUC, and pENTR/NotI_AscI. The 5’-part-LUC fragment for each construct consisted of a PCR amplification product from the start codon to the insertion site of the LUC+ gene in pENTR_LUC+. A common forward primer (5’-GCAGGCTCCGCGGCCGCCATG-3’) and a reverse primer specific for each construct were used for amplification (Supplementary Table S2). Each 3’-part-LUC fragment was a PCR amplification product from the insertion site to the stop codon of the LUC+ gene in pENTR_LUC+. A common reverse primer (5’-AAGCTGGGTCGGCGCGCCTTA-3’) and a forward primer specific for each construct were used for the PCR amplification (Supplementary Table S2). pENTR/NotI_AscI was linearized pENTR_MCS fragment obtained through digestion with AscI and NotI (New England Biolabs, MA, USA). pENTR_MCS had the multiple cloning site, including AscI and NotI, at the cloning site of pENTR-D-TOPO.
A set of the three DNA fragments was integrated into each construct by the In-fusion method (In-Fusion^®^ Snap Assembly Master Mix; Takara Bio). The CaMV35S promoter sequence was then cloned into the pENTR5’-TOPO cloning vector (Thermo Fisher Scientific). Finally, pENTR_LUCXAAIns24bp/26bp (attL1-attL2) fragments were individually integrated with pENTR5’_CaMV35S (attL4-attR1) into a pUC18-based destination vector (attR4-attR2), carrying a Nos terminator, through LR reactions (Gateway LR Clonase II Enzyme mix; Thermo Fisher Scientific)^28^. Similarly, the AtCCA1 promoter sequence was cloned into the pENTR5’-TOPO cloning vector^4^, and pENTR_LUC40Ins24bp/26bp (attL1-attL2) fragments were individually integrated with pENTR5’_AtCCA1 (attL4-attR1) into a pUC18-based destination vector (attR4-attR2), carrying a Nos terminator, through LR reactions.
pENTR_AtU6-26::sgRNA-LUCX AA was constructed as follows: The DNA fragment comprising the 20-bp target region was made by annealing fragments 5’-attgGTTCTACCTATGATTCCCAG-3’ and 5’-aaacCTGGGAATCATAGGTAGAAC-3’. This DNA fragment was cloned into the BbsI-site of the pENTR_AtU6-26::sgRNA vector^30^.
pR4GWB501_CaMV35S::LUC40Ins26bp was constructed by integrating pENTR_LUC40Ins26bp, pENTR5’_CaMV35S, and pR4GWB501 by LR reaction. pR4GWB501 is a binary transformation vector harboring a hygromycin-resistant selectable marker^61^ (Supplementary Fig. 11).
Production of Transgenic lines
Stable transformation of Arabidopsis thaliana (Col-0) with Agrobacterium tumefaciens (EHA105 strain) carrying pR4GWB501_CaMV35S::LUC40Ins26bp was performed using the floral dip method^62^. T1 seeds were sown on 0.5× MS agar medium containing 20 mg/mL hygromycin B (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) and 200 mg/mL cefotaxime sodium (Claforan^®^; Sanofi, Paris, France). The 41 lines of seedlings that survived in the selection medium were transplanted into soil and grown to obtain T2 seeds. The T2 seeds from each plant were sown on the same selection agar medium. Based on the segregation of hygromycin B resistance, 13 lines were identified as single-locus transformants. Among these lines, we selected those T2 generation lines that showed high expression levels of LUC40Ins26bp. Homozygous lines (T2 generation) were selected based on the segregation of hygromycin B resistance in the T3 generation. The T3 seedlings of these homozygous lines were used for bioluminescence experiments.
Particle bombardment experiments
Reporter constructs or CRISPR/Cas9 constructs were introduced into duckweed or Arabidopsis using a particle bombardment system (PDS-1000/He; Bio-Rad, Hercules, CA, USA) as previously described^30,48^. Gold particles were prewashed and stocked in 50% glycerol (duckweed, 1-µm diameter, 60 mg/mL; Arabidopsis, 0.6-µm diameter, 13 mg/mL). The number of gold particles per unit volume was equalized. For Arabidopsis and duckweed, gold particles (0.104 mg and 0.480 mg, respectively) were coated with plasmid DNA. The concentrations and amounts of constructs coated on the gold particles in each experiment are shown in Supplementary Table S3. After gene transfection, plants were immediately transferred to 60-mm petri dishes (IWAKI, Tokyo, Japan) containing 8 mL NF medium containing 8 µL of 0.1 mM D-luciferin.
Bioluminescence monitoring and quantification
Bioluminescence monitoring at the single-cell level was performed as previously described with minor modifications^22,28^. A lens (XENON 0.95/22MM C-mount; Schneider Optics, Rueil-Malmaison, France) attached to a short-pass filter (SV630; Asahi Spectra, Tokyo, Japan) to reduce delayed autofluorescence from the chloroplasts was used together with a bioluminescence imaging system consisting of an EM-CCD camera (ImagEM X2 C9100-23B for snapshot imaging or ImagEM C9100-13 for time-lapse imaging; Hamamatsu Photonics, Shizuoka, Japan). Extension rings of 5 and 16.5 mm thick were used for whole-dish and close-up imaging, respectively. HC-image (for snapshot imaging) or HOKAWO (for time-lapse imaging) software (Hamamatsu Photonics) was used to control the imaging system. Bioluminescence images (16-bit TIFF format) were captured twice at several exposure times with the ImagEM camera (cooled at − 80 °C) at an EM gain of 1200 after at least a 4-min dark treatment for autofluorescence decay. Image analysis and post-processing were performed using ImageJ software (LOCI, University of Wisconsin, USA). To remove the cosmic-ray spike, the minimum value for each pixel between two sequential images was used for analysis. The signal intensity of the bioluminescent spot was quantified as an integrated density of the region of interest (ROI; 6 × 6 pixels). The ROIs that could be determined from a single cell were manually selected for each image. To quantify the background noise, we selected ten ROIs (10 × 10 pixels) in the plant area without bioluminescent spots and calculated the median pixel signal intensity of each ROI. We then defined the mean value of the medians of the ten ROIs as the background value for each pixel. The background level varied among samples in a range from 1920 to 2300. The bioluminescence intensities were calculated using the following formula^27^:
Number of photons per min = (signal intensity − background value) × Conversion factor / (Analog gain × EM gain × Conversion efficiency) × 60 (sec) / Exposure time (sec) = (signal intensity – background value) × 5.8 / (1 × 1200 × 0.9) × 60 (sec) / Exposure time (sec).
RNA isolation and qPCR analysis
Total RNA was isolated from five 14-day-old Arabidopsis seedlings, grown under constant light conditions, using a NucleoSpin^®^ RNA kit (Takara Bio). cDNA was synthesized using ReverTra Ace (TOYOBO). qPCR was performed using a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) with the THUNDERBIRD SYBR qPCR Mix (TOYOBO). cDNA levels were normalized to those of the housekeeping control gene, ISOPENTENYL PYROPHOSPHATE: DIMETHYLALLYL PYROPHOSPHATE ISOMERASE 2 (AtIPP2). The following primers were used: 5’-TCAGGTGGCTCCCGCTGAATTG-3’ and 5’-CCGTCATCGTCTTTCCGTGCTC-3’ for LUC40Ins26bp, and 5’-GTATGAGTTGCTTCTCCAGCAAAG-3’ and 5’-GAGGATGGCTGCAACAAGTGT-3’ for AtIPP2.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 1
Supplementary Material 2
Supplementary Material 3
Supplementary Material 4
Supplementary Material 5
Supplementary Material 6
Supplementary Material 7
Supplementary Material 8
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nishiguchi, T. et al. Development of red-shifted mutants derived from luciferase of Brazilian click beetle Pyrearinus termitilluminans. J. Biomed. Opt.2010.1117/1.JBO.20.10.101205 (2015).10.1117/1.JBO.20.10.10120526313214 · doi ↗ · pubmed ↗
- 2Mc Vey, M., La Rocque, J. R., Adams, M. D. & Sekelsky, J. J. Formation of deletions during double-strand break repair in Drosophila Dm Blm mutants occurs after strand invasion. Proc. Natl. Acad. Sci. USA 101, 15694–15699 (2004).10.1073/pnas.0406157101 PMC 52485115501916 · doi ↗ · pubmed ↗
- 3Tuladhar, R. et al. CRISPR-Cas 9-based mutagenesis frequently provokes on-target m RNA misregulation. Nat. Commun.1010.1038/s 41467-019-12028-5 (2019).10.1038/s 41467-019-12028-5PMC 673129131492834 · doi ↗ · pubmed ↗
- 4Feng, Z. et al. Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc. Natl. Acad. Sci. USA 111, 4632–4637 (2014).10.1073/pnas.1400822111 PMC 397050424550464 · doi ↗ · pubmed ↗