Long-standing myalgia and hypouricemia in a young woman: Case report and review of the literature
Ruta Venyte, Corina Schuster-Amft, Frank Behrendt, Nicole Nyfeler, Katrin Parmar, Johannes Lorscheider, Leo H. Bonati, Ulrich A. Walker, Hans Ulrich Gerth

TL;DR
A 46-year-old woman with long-term muscle pain and low uric acid levels was diagnosed with xanthinuria, a rare inherited metabolic disorder.
Contribution
The paper presents a rare case of xanthinuria with overlapping symptoms of fibromyalgia, highlighting diagnostic challenges.
Findings
The patient exhibited hypouricemia and elevated urinary xanthine levels, consistent with xanthinuria.
Sonographic findings revealed small kidney stones, supporting the xanthinuria diagnosis.
Clinical symptoms overlapped with fibromyalgia, making differential diagnosis difficult.
Abstract
Eine 46-jährige Patientin leidet seit dem 27. Lebensjahr an multiplen Symptomen wie Arthralgien, Myalgien, allgemeiner Müdigkeit, Erschöpfung, Konzentrationsstörungen, Vergesslichkeit, Ein- sowie Durchschlafstörungen und Depression. Rheumatologisch wurden vorbefundlich eine rheumatoide Arthritis diagnostiziert mit fehlendem Ansprechen auf eine Basistherapie sowie ebenfalls eine sekundäre Fibromyalgie. Bei laborchemisch Hypourikämie mit nicht nachweisbarer Harnsäure im Serum erfolgten ergänzende Stoffwechseluntersuchungen, die massiv erhöhte Xanthinwerte im Urin zeigten. Bei normwertiger Nierenfunktion und unauffälligem Urinsediment konnten sonographisch kleine Konkremente im Nierenbeckenkelchsystem beidseits dargestellt werden. Es konnte die Diagnose einer Xanthinurie gestellt werden. Hierbei handelt es sich um eine seltene autosomal-rezessiv vererbte Störung des Purinstoffwechsels mit…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1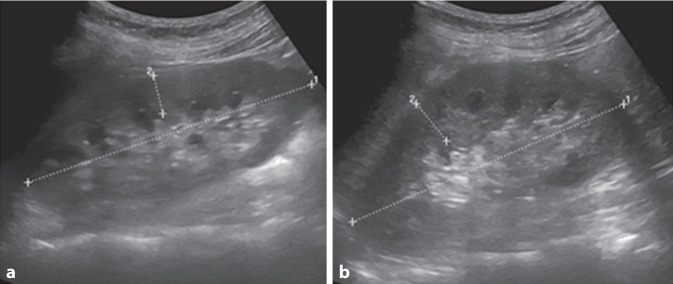 Figure 2
Figure 2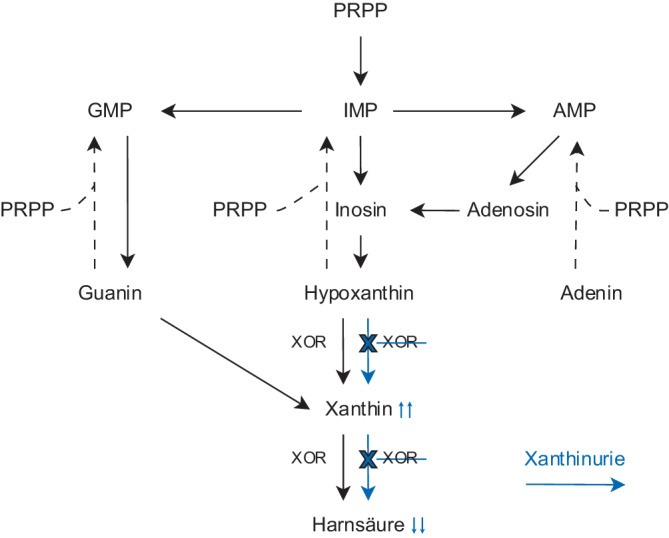 Figure 3
Figure 3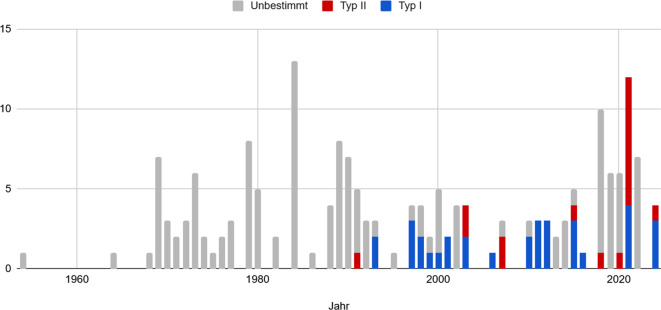 Figure 4
Figure 4 Figure 5
Figure 5- —Universität Münster (1056)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGout, Hyperuricemia, Uric Acid · Fibromyalgia and Chronic Fatigue Syndrome Research · Alcoholism and Thiamine Deficiency
Anamnese
Eine 46-jährige Patientin leidet seit dem 27. Lebensjahr an Arthralgien, Myalgien, allgemeiner Müdigkeit, Erschöpfung, Konzentrationsstörungen, Vergesslichkeit, Ein- sowie Durchschlafstörungen und Depression. Im Jahr 2020 wurde eine seronegative rheumatoide Arthritis diagnostiziert bei sonographisch Ergüssen in multiplen Kleinfingergelenken beidseits mit Power-Doppler-Aktivität. Eine Basistherapie mit Methotrexat und Prednison wurde begonnen und nach wenigen Monaten bei fehlender Wirkung auf die Arthralgien sowie gastrointestinaler Symptomatik beendet. Die Arthritiden rezidivierten im Verlauf auch nicht, sodass retrospektiv die Diagnose der rheumatoiden Arthritis infrage gestellt werden kann. Im Verlauf wurde die Diagnose einer sekundären Fibromyalgie bei Druckschmerzhaftigkeit aller ACR(American College of Rheumatology)-Tenderpoints (18/18) in der klinischen Untersuchung gestellt. Alle multimodalen Therapieversuche (Homöopathie, Akupunktur, Schröpfen, Physiotherapie, Wassertherapie, medizinische Trainingstherapie, Analgetika) brachten keine dauerhafte Linderung.
Befund
Die Patientin stellte sich in der rheumatologischen Sprechstunde zur Zweitmeinung in sehr gutem Allgemeinzustand und normalem Ernährungszustand vor. Der rheumatologische Status war insgesamt unauffällig bis auf eine diffuse Berührungsempfindlichkeit der Haut, eine allgemeine nicht gelenkbezogene Druckschmerzhaftigkeit sowie paravertebrale Myogelosen. Insbesondere zeigten sich keine Arthritis und eine unauffällige Textur der Muskulatur ohne Vaskulitis- oder Kollagenosestigmata. Vorbefundlich dokumentiert sind negative Rheumafaktoren, antinukleäre Antikörper (ANA), extrahierbare nukleäre Antigene (ENA), CPP(antizyklisches citrulliniertes Peptid)-Antikörper sowie antineutrophile zytoplasmatische Antikörper (ANCA) (Tab. 1), zudem bestand keine humoral entzündliche Reaktion. Bei laborchemisch Hypourikämie mit fehlendem Nachweis von Harnsäure im Serum und im Urin erfolgten ergänzende Stoffwechseluntersuchungen, die massiv erhöhte Xanthinwerte zeigten (Tab. 2).Tab. 1Rheumatologisches Basis-Labor im SerumAnalyseResultatReferenzHämoglobin144 g/l120–160Thrombozyten190 G/l150–450Leukozyten6,3 G/l4,5–11,5Senkung (EDTA)2 mm/h< 30Natrium138 mmol/l136–145Kalium4,5 mmol/l3,5–5,1Calcium2,31 mmol/l2,15–2,50Magnesium0,88 mmol/l0,66–1,07Phosphat1,24 mmol/l0,81–1,45Harnstoff4,0 mmol/l2,1–7,1Kreatinin60 µmol/l40–80eGFR (Krea, CKD-EPI 2009)106 ml/min/1,73 m^2^> 60C‑reaktives Protein< 0,5 mg/l< 10AST (GOT)29 U/l5–35Gamma-GT10 U/l< 38Alkalische Phosphatase63 U/l46–116Harnsäure< 20 µmol/l137–357Vitamin D, 25-OH57 nmol/l50–220CK gesamt108 U/l34–145Parathormon29,1 ng/l18,4–80,1TSH0,93 mU/l0,55–4,78Rheumafaktor< 4 kIU/l< 14CCP IgG< 0,5 kU/l< 5ANA IIF HEp‑2< 160< 160Tab. 2Urinbefund und ergänzende Stoffwechselparameter im UrinAnalyseResultatReferenzLeukozytenNegativNegativErythrozytenNegativNegativProteinNormalNormalKreatinin2,4 mmol/l–Harnsäure< 2,7 mmol/mol Kreatinin229–571Xanthin177 mmol/mol Kreatinin< 5,0Hypoxanthin13 mmol/mol Kreatinin< 13Inosin< 0,2 mmol/mol Kreatinin< 0,8Dihydrouracil4,6 mmol/mol Kreatininn. d.
Nativradiologisch bestanden keine typischen arthritischen Veränderungen der Hände und Füße noch Zeichen einer Kristallarthropathie der Knie und Hände (Abb. 1). Bei normwertiger Nierenfunktion und unauffälligem Urinsediment konnten in der Abdomensonographie kleine Konkremente im Nierenbeckenkelchsystem (Nierengrieß) beidseits ohne Harnstauung dargestellt werden (Abb. 2). Zudem ergab sich elektroneurographisch kein Hinweis auf ein Karpaltunnelsyndrom bei ebenfalls unauffälligem Befund der Magnetresonanztomographie der Lendenwirbelsäule.Abb. 1Konventionelle Röntgenaufnahmen der a Hände und b Füße beidseits mit unspezifischen Veränderungen. An den Knien (c links und d rechts) beidseits keine Meniskusverkalkung als Hinweis auf eine Kalziumpyrophosphat-Dihydrat(CPPD)-ArthropathieAbb. 2Sonographisch orthotope Lage und regelhafte Größe der Nieren beidseits, a rechts 11,6 cm lang, b links 11 cm, mit beidseits regelhafter Parenchymdicke, a rechts 1,59 cm, b links 1,91 cm, und kleine Konkremente im Nierenbeckenkelchsystem (Nierengrieß) beidseits ohne Harnstauung
Diagnose
Anhand der fehlenden Harnsäure (im Serum und Urin) sowie der erhöhten Xanthin-Spiegel im Urin konnte die Diagnose einer Xanthinurie gestellt werden [1]. Hierbei handelt es sich um eine seltene autosomal-rezessiv vererbte Störung des Purinstoffwechsels, bei der das Enzym Xanthinoxidreduktase (XOR) (Enzym mit beiden Formen Xanthindehydrogenase [XDH] und Xanthinoxidase [XO]) defizitär ist [2].
Biochemische Eigenschaften, Einteilung und Genetik
Die XOR katalysiert die Umwandlung von Hypoxanthin zu Xanthin und von Xanthin zu Harnsäure. Xanthin wird zudem auch aus Guanin gebildet. Die Akkumulation von Hypoxanthin wird größtenteils durch den Salvage Pathway mit 5‑phospho-α-D-ribosyl-1-pyrophosphat (PRPP) als Ko-Substrat in Inosinmonophosphat umgewandelt (Abb. 3; [1, 2]).Abb. 3Vereinfachte Darstellung des Purinstoffwechsel physiologisch (schwarz) sowie bei Xanthinurie (blau): XOR katalysiert die Bildung von Xanthin aus Hypoxanthin und von Harnsäure aus Xanthin. Xanthin wird ebenfalls aus Guanin gebildet. Bei der klassischen Xanthinurie akkumuliert aufgrund der fehlenden Funktion der XOR Hypoxanthin und Xanthin, wobei Hypoxanthin ebenfalls über den Salvage Pathway zurückgebildet wird zu Inosinmonophosphat (IMP)
Die klassische Xanthinurie wird in 2 Typen eingeteilt während zwischenzeitlich pro Typ mehrere Mutationen beschrieben wurden. Bei Typ 1, in Online Mendelian Inheritance in Man (OMIM), einer medizinischen Online-Datenbank menschlicher Gene sowie genetischer Phänotypen und Erkrankungen als Nummer 278300 bezeichnet, handelt es sich um einen genetischen Defekt der XOR im Chromosom 2p23.1, daher fehlt isoliert die Funktion von XOR. Beim Typ 2 (OMIM 603592) betrifft der genetische Defekt die Molybdän-Kofaktor-Sulfurase (MOCOS) im Chromosom 18q12.2. Diese Mutation verhindert sowohl die Aktivierung der XOR als auch der Aldehydoxidase (AO), was ebenfalls zu einer Akkumulation von Xanthin führt [1, 3, 4].
Der ergänzende Molybdän-Kofaktor-Mangel wird als Xanthinurie Typ 3 (OMIM 252150) bezeichnet, der neben dem Verlust der Enzymaktivität von XOR und AO auch die Sulfitoxidase (SO) betrifft. Da hierbei jedoch betroffene Patienten eine hohe Symptomlast haben (neurologische Ausfälle, Krämpfe, Muskeltonusstörungen, Entwicklungsstörungen, Linsenluxation) und die meisten im ersten Lebensjahr versterben [5, 6], ist dieses in unserem Fall klinisch unwahrscheinlich.
Obwohl die genetischen Defekte verschiedene Gene betreffen, sind die beiden Varianten der klassischen Xanthinurie, Typ I und Typ II, klinisch nicht zu differenzieren [7]. Die Unterscheidung zwischen Typ 1 und Typ 2 kann mit dem Allopurinol-Loading-Test erfolgen, bei dem Oxypurinol (Abbauprodukt aus Allopurinol) im Serum oder Urin nach Verabreichung von Allopurinol gemessen wird. Wird Allopurinol verabreicht, aber nicht zu Oxypurinol metabolisiert, liegt eine Xanthinurie vom Typ 2 vor, bei der sowohl die XOR als auch die AO fehlen, während bei einer Metabolisierung zu Oxypurinol eine Xanthinurie vom Typ 1 vorliegt [1]. Die Diagnose einer Xanthinurie kann zudem durch eine niedrige XDH-Aktivität in der Leber oder der Duodenalschleimhaut mittels Biopsie bestätigt werden [3, 8]. Klinisch ist dies jedoch nicht relevant, da die Therapie und Prognose sich nicht zwischen beiden Xanthinurietypen unterscheiden.
Die Xanthinurie wird autosomal-rezessiv vererbt [1]. Die Großmütter unserer Patientin waren Cousinen, was eine Wahrscheinlichkeit einer Homozygote bei Nachkömmlingen erhöht. Die erweiterte Familienanamnese ergab ebenfalls Nierensteinleiden bei Verwandten (Vater und Großvater väterlicherseits sowie mütterlicherseits). Lebende Verwandte (2 Brüder, die Schwester und 3 Kinder unserer Patientin) sind bisher asymptomatisch mit normwertiger Harnsäure im Serum. Eine genetische Untersuchung wurde nicht durchgeführt.
Epidemiologie und Literatur-Review
Der erste Fall einer Xanthinurie wurde von Dent und Philpot im Jahr 1954 beschrieben [9]. Weltweit sind bisher nur wenige Fallberichte von Patienten mit Xanthinurie veröffentlicht. Die Inzidenzschätzung liegt zwischen 1:100.000 und 1:1.000.000, aber manche Autoren berichten, dass die Erkrankung stark unterdiagnostiziert sei [10]. Eine systematische Literaturrecherche aller Fallberichte in PubMed ergab 104 Originalarbeiten mit 186 Patienten (s. elektronisches Zusatzmaterial online). Hierbei wird oft nicht sicher zwischen Xanthinurie Typ 1 und 2 differenziert (Abb. 4). Weniger frühe veröffentlichte Fallberichte spezifizieren zudem nicht, ob es sich um eine Neubeschreibung oder ein Follow-up handelt. Die Anzahl der veröffentlichten Fälle zeigt jedoch über die Jahre keine Veränderung.Abb. 4Publizierte Fallberichte der Xanthinurie im zeitlichen Verlauf bis 2024 (blau Typ I vs. rot Typ II vs. grau unbestimmt)
Symptome
Die meisten Patienten mit Xanthinurie Typ I und II sind asymptomatisch, mögliche klinische Symptome sind jedoch Urolithiasis aufgrund erhöhter Xanthinausscheidung und Myalgien aufgrund von intramuskulären Xanthinablagerungen. Als weitere Komplikationen wurden Arthralgien und Niereninsuffizienz im Verlauf der Erkrankung beschrieben [1, 11, 12]. Klinisch stehen in unserem Fall neben der Nephrolithiasis die typischen Symptome der „Fibromyalgie“ (chronische generalisierte muskuloskeletale Schmerzen, Fatiguesymptomatik, Schlafstörungen sowie kognitive Einschränkungen und depressive Symptomatik [13–16]) im Vordergrund, die sich teilweise mit den möglichen Symptomen einer Xanthinurie überlappen (Tab. 3). Es kann klinisch nicht sicher differenziert werden, ob es sich bei den Symptomen um 2 unabhängige Erkrankungen handelt oder um eine sekundäre Fibromyalgie als Folge der Xanthinurie.Tab. 3Typische Symptome Fibromyalgie [13–16] und Xanthinurie [1, 11, 12]Fibromyalgie (%)Xanthinurie (%)Myalgien (~ 45)Myalgien (~ 6,9)Arthralgien (~ 45)Arthralgien (~ 5,1)Fatigue (~ 40 %)Urolithiasis (~ 30)Kognitionsstörung (~ 40)–Depression (~ 40)–Schlafstörung (~ 30)–
Therapie und Verlauf
Xanthin ist in Wasser schwer löslich, und die Löslichkeit wird durch den pH-Wert nur geringfügig beeinflusst, sodass eine Alkalisierung des Urins nicht wirksam ist, um die Steinbildung zu verhindern. Die einzige Prophylaxe besteht in einer erhöhten Diurese (vermehrte Dilutation durch gesteigerte Flüssigkeitszufuhr) und einer purinarmen Diät (verminderte Steinbildung durch Substratmangel). Kontraintuitiv gleichen sich somit die alimentäre Therapie der Hypo- und Hyperurikämie. Im Verlauf ist insbesondere die interdisziplinäre nephrologische Mitbetreuung notwendig, da die Entwicklung einer Niereninsuffizienz möglich ist [17–20].
Fazit für die Praxis
Auffälligen Befunden sollte auch dann nachgegangen werden, wenn eine andere Erkrankung wie Fibromyalgie ausreichend diagnostiziert werden kann. Bei Patienten mit unklarer Hypourikämie sind detaillierte Untersuchungen der Purinstoffwechselparameter erforderlich, da eine frühzeitige Diagnose und Behandlung mit hoher Flüssigkeitszufuhr und Ernährungsumstellung Komplikationen wie Nierensteine reduzieren können.
Supplementary Information
Systematische Literatur-Recherche aller Fallberichte in PubMed