The complete chloroplast genome of Tinospora sinensis and its phylogenetic analysis
Jiahua Chen, Yang Ni, Guoan Shen, Chang Liu

TL;DR
This paper presents the full chloroplast genome of Tinospora sinensis, a medicinal plant, and explores its evolutionary relationships.
Contribution
The first complete chloroplast genome assembly and phylogenetic analysis of Tinospora sinensis is reported.
Findings
The chloroplast genome is 160,664 bp long with 126 genes and a GC content of 39.31%.
256 SSRs and 50 dispersed repeats were identified as potential molecular markers.
Phylogenetic analysis confirmed close relationships with other Tinospora species.
Abstract
Tinospora sinensis (Lour.) Merr. (Menispermaceae) is a medicinal plant with limited genomic resources available. Here, we assembled and annotated its 160,664 bp chloroplast genome, identifying 126 genes with a GC content of 39.31%. We detected 256 SSRs and 50 dispersed repeats, which represent potential molecular markers. IR boundary analysis showed slight expansions/contractions involving rps19, ndhF, and ycf1. Phylogenetic analysis confirmed its close evolutionary relationship with other Tinospora species. This is the first comprehensive genomic characterization of T. sinensis, providing valuable resources for taxonomy, evolution, and medicinal research in Menispermaceae.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1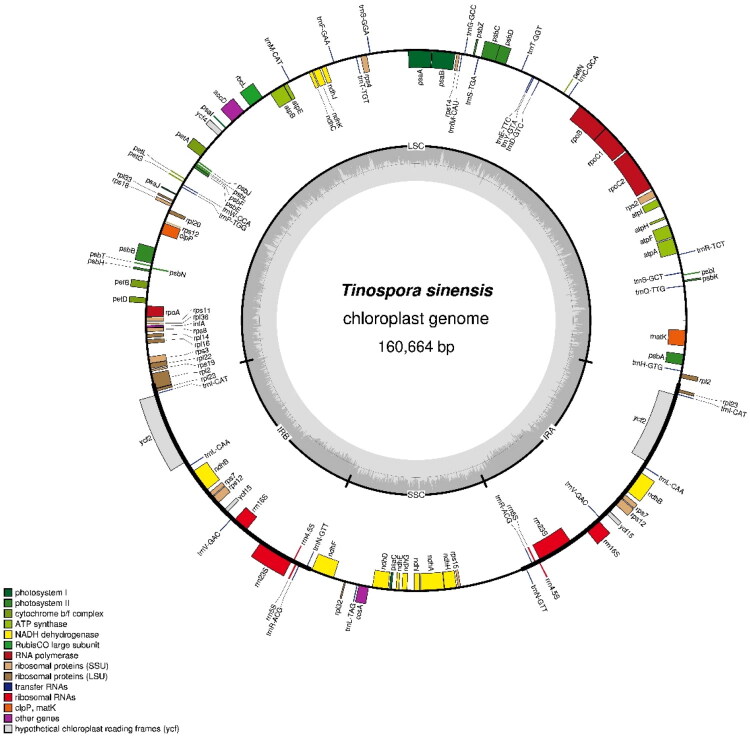 Figure 2
Figure 2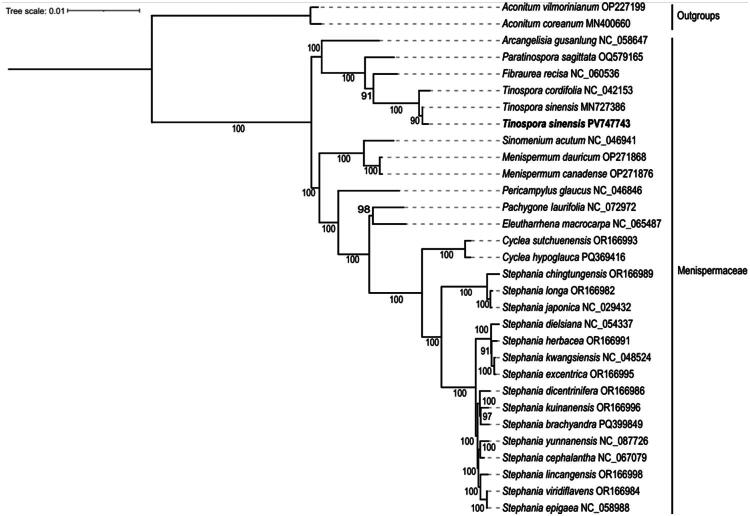 Figure 3
Figure 3- —Guangxi Laibin City Science Research and Technology Development Plan
- —National Science & Technology Fundamental Resources Investigation Program of China
- —Hunan Provincial Special Project for Construction of Innovation Demonstration Area at Chenzhou City under National Sustainable Development Plan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhytochemicals and Medicinal Plants · Medicinal Plants and Bioactive Compounds · Biological and pharmacological studies of plants
Introduction
Tinospora sinensis (Lour.) Merr. 1934 also known as Qingjiuniu in Chinese, is a climbing plant of the genus Tinospora of the family Menispermaceae. It is widely distributed across India, China, Northwestern South Africa, Myanmar, and Sri Lanka. Previous studies have revealed close genetic similarities among the three Tinospora species found in India: T. sinensis, T. cordifolia, and T. crispa (Liang et al. 2024). The stems of T. sinensis are commonly used as traditional herbal medicines. Recent studies have identified 57 chemical compounds from T. sinensis, including three newly identified structures belonging to lignans, alkaloids, and phenolic compounds (Lam et al. 2018; Liang et al. 2024). Pharmacological investigations have demonstrated its antioxidant, anti-neuroinflammatory, and antidiabetic properties (Banerjee et al. 2020; Zhou et al. 2020). In addition, Tinospora species are widely used as components in various traditional formulations (Upadhyay et al. 2010; Lam et al. 2012). While a chloroplast genome sequence (MN727386) of T. sinensis is available in GenBank of NCBI, the absence of a formal publication and detailed characterization limits its utility. Therefore, there is an urgent need to clarify the phylogenetic relationships of T. sinensis and its closely related species, and to identify reliable molecular markers to support bioprospecting and quality assurance of T. sinensis products. This study provides the first comprehensive analysis of the T. sinensis chloroplast genome to address these gaps.
Chloroplasts are double-membraned organelles found in plants, algae, and certain protists. In green plants, they serve as the primary organelles for photosynthesis and possess their own genome. Chloroplast genomes typically range from 120 to 160 kb in size and exhibit a highly conserved quadripartite structure (Lu et al. 2023; Zhang et al. 2023). The chloroplast genomes consist of a single circular DNA molecule composed of two inverted repeat (IR) regions, which separate the large single-copy (LSC) region from the small single-copy (SSC) region (Daniell et al. 2016). With the advancement of high-throughput sequencing technologies, chloroplast genomes have been increasingly applied in studying plant systematics and taxonomy. They have become important molecular tools for investigating plant genetic diversity and evolutionary relationships. To date, one T. sinensis chloroplast genome sequence has been submitted to the public database (MN727386). However, no formal publication associated with it could be found.
In this study, we assembled and annotated the complete chloroplast genome of T. sinensis using high-throughput sequencing technology, aiming to characterize its structural features and determine the phylogenetic relationship of T. sinensis and its congeneric species. This research provides essential molecular data to support the conservation and utilization of T. sinensis, and contributes valuable genomic resources for the phylogenetic and evolutionary study of Menispermaceae.
Materials and methods
Fresh leaves from a single individual were collected from Tongmu Township, Jinxiu Yao Autonomous County, Laibin City, Guangxi Zhuang Autonomous Region, China (latitude 24.191745°, longitude 109.988645°; altitude: 130 m) and immediately preserved in liquid nitrogen. Sampling was authorized by Xinmin Pan ([email protected]). The voucher specimen (No. JXHC015) has been deposited in the herbarium of the Medicinal Plant Garden, Institute of Medicinal Plant Development (Figure 1).
The leaves of T. sinensis, photographed by Jiahua Chen in Jinxiu Yao Autonomous County, Guangxi, China. The collection information, added automatically at the time of photographing, are shown in the left lower corner. The translation was shown in square bracket. The features of the plant are the following: leaves broadly ovate to suborbicular, entire-margined, apex acute, base deeply cordate or slightly cordate, both surfaces covered with soft hairs.
Total DNA was extracted using the Plant Genomic DNA Kit, whole-genome sequencing was performed on the PacBio Sequel II platform, generating ∼10 Gb of high-fidelity (HiFi) long-read data. The long-read data were used for de novo assembly of the complete plastid genome using the oatk assembler (v1.0) (Kolmogorov et al. 2019).
To verify the structural integrity and accuracy of the assembly, the resulting genome was visualized using Bandage v0.8.1 (Wick et al. 2015). In addition, we mapped the reads to the assemblies to determine how well the assemblies were supported by the reads (Ni et al. 2023). The genome was annotated using GeSeq (Tillich et al. 2017) and CPGAVAS2 (Shi et al. 2019). The correctness of the annotation results was examined using CPGView (Liu et al. 2023). All genomic features, including cis-spliced and trans-spliced genes were also visualized by CPGview, whereas circular genome mapping was generated by the GeSeq online website tool (https://chlorobox.mpimp-golm.mpg.de/geseq.html).
To quantitatively evaluate the preferential usage of individual codons among synonymous alternatives, the Relative Synonymous Codon Usage (RSCU) metric has been widely adopted in codon usage studies. In this study, RSCU values were calculated using the following formula (Choudhuri and Sau 2024):
Interspersed repeat sequences were identified using the online tool REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer), with the parameters set to a maximum of 50 repeats and a minimum repeat size of 8 bp (Kurtz et al. 2001). Four types of repeats were detected: forward, palindromic, reverse, and complement repeats. Simple sequence repeats (SSRs), also known as microsatellites, are short tandemly repeated DNA sequences widely distributed throughout the genome. They typically consist of repeat motifs ranging from 2 to 6 nucleotides in length, repeated in tandem multiple times (Alves et al. 2023). MISA (Microsatellite Identification Tool) was used for the prediction of SSRs (Beier et al. 2017), with parameter settings as follows: copy numbers of single-base repeats > 8, double-base repeats > 5, triple-base repeats > 3, quadruple-base repeats > 3, quintuple-base repeats > 3, and hexa-base repeats > 3.
To visualize the junctions between the inverted repeat (IR), large single-copy (LSC), and small single-copy (SSC) regions of the chloroplast genomes, we employed IRscope (Amiryousefi et al. 2018).
For phylogenetic analysis, complete chloroplast or plastid genome sequences of related species from different genera of the same family were downloaded from the GenBank database and the target species were processed using PhyloSuite (v1.2.3) (Zhang et al. 2019). The conserved genes common to all species were extracted using PhyloSuite and the maximum likelihood phylogenetic tree was reconstructed by running UFBS (UltraFast BootStraps) 1000 times using IQ-TREE and cpREV substitution models. The generated trees were visualized using the Interactive Tree of Life (iTOL) platform (https://itol.embl.de/) (Letunic and Bork 2024).
Results
We obtained ∼21 Gb of raw sequencing data. The length of the assembled chloroplast genome was 160,664 bp, and the coverage depth ranged from a minimum of 128× to a maximum of 4405× (Figure S1) suggesting that the assembly is strongly supported with the sequencing reads. The high sequencing depth and coverage indicate that the genome assembly is highly reliable and suitable for subsequent structural and comparative analyses.
The plastome has a typical quadripartite structure, including a large single-copy (LSC) region of 92,353 bp, a small single-copy (SSC) region of 20,435 bp, and two inverted repeats (IR) regions of 23,938 bp each. The average GC content was 39.31%.
A total of 126 functional genes are annotated, including 82 protein-coding genes (PCGs), 36 transfer RNA (tRNA) genes, and 8 ribosomal RNA (rRNA) genes (Figure 2). A total of 9 PCGs [atpF, rpoC1, petB, petD, rpl16, rpl2, ndhB (2x), and ndhA] contained one intron, while 4 genes [ycf3, clpP, and rps12 (2x)] contained two introns (Figure S2). In addition, 7 tRNA genes [trnK-UUU, trnT-CGU, trnL-UAA, trnE-UUC (2x), trnA-UGC (2x)] also contained one intron (Figure S2). Furthermore, rps12 gene was trans-spliced and had three independent exons, two of which were duplicated in the IR region (Figure S3).
Circular gene map of the chloroplast genome of T. sinensis. The genome has a typical quadripartite structure, consisting of a large single-copy (LSC) region, a small single-copy (SSC) region, and a pair of inverted repeats (IRA and IRB), totaling 160,664 bp. Genes are color-coded according to functional categories, including genes involved in photosynthesis (photosystem I and II, ATP synthase, cytochrome b6f complex, and NADH dehydrogenase), transcription and translation (RNA polymerase, ribosomal proteins, rRNAs, tRNAs), and other functions. Genes transcribed clockwise are located on the outer side, and those transcribed counterclockwise are on the inner side of the circle.
Then we estimated the relative synonymous codon usage (RSCU) and codon usage frequency. In the chloroplast genome of T. sinensis, a total of 64 codons were identified, including three stop codons. The codon–anticodon recognition pattern revealed that among the stop codons, UAA was the most frequently used. In addition, GCU for alanine, AGA for arginine, UUA for leucine, UCU for serine, and ACU for threonine exhibited relatively high usage frequencies (Figure S4).
Among all codons, 31 codons showed RSCU values >1, of which 29 (93.54%) ended with either A or U. Conversely, among the 31 codons with RSCU values <1, 28 (90.32%) ended with either C or G. These findings indicate a strong codon usage bias toward codons ending in A or U in this chloroplast genome.
Repeat sequence analysis using the REPuter software identified a total of 50 dispersed repeats, including 21 forward repeats, 7 reverse repeats, 19 palindromic repeats, and 3 inverted repeats.
Their distribution patterns provide insights into genome stability, sequence evolution, and potential functional roles, serving as a molecular basis for investigating genomic evolutionary mechanisms. The presence of numerous palindromic and forward repeats suggests a potential role in promoting sequence rearrangements and plastome evolution in T. sinensis (Figure S5).
In the chloroplast genome of T. sinensis, a total of 256 SSRs were identified. Among these, mononucleotide repeats were the most abundant (140, 54.69%), followed by trinucleotide repeats (85, 33.20%), tetranucleotide repeats (17, 6.64%), dinucleotide repeats (12, 4.69%), and a small number of pentanucleotide and hexanucleotide repeats (each 1, 0.39%). Most mononucleotide SSRs were composed of A/T motifs, consistent with the AT-rich nature of chloroplast genomes (Figure S6). The abundance and distribution of these SSRs provide valuable resources for future population genetic and evolutionary studies in Tinospora species.
Comparative analysis of the chloroplast genome between our newly assembled (PV747743) and the released one (MN727386.1) revealed high sequence similarity and conserved genome structure. Overall, the alignment covered nearly the entire genome with >99.9% identity, indicating strong conservation at the genomic level. Despite this overall conservation, structural variations were detected at the boundaries of the LSC, SSC, and IR regions, prompting a detailed comparison of junction features.
The boundaries of the LSC, SSC, and IR regions are analyzed by comparing the newly assembled chloroplast genome of T. sinensis (PV747734) with the reference genome from the GenBank database (MN727386). The lengths of the LSC, SSC, and IR regions differed between the two genomes, reflecting potential structural variation. Notably, the IR/LSC junction in T. sinensis (PV747734) involves the rpl2 gene. Such boundary variation may affect the copy number or expression of rpl2, which could in turn influence chloroplast ribosomal function. The biological significance of this structural difference is further discussed in the Discussion section (Figure S7).
In the newly assembled genome, the rpl2 gene was found spanning the LSC/IRb junction, with 1466 bp of the gene located upstream within the IRb region. The ndhF gene was situated 61 bp upstream of the SSC/IRb border, while rpl23 was located 536 bp downstream of the IRa region.
In contrast, the reference genome (MN727386) exhibited different boundary arrangements. The rps19 gene was positioned at the LSC/IRb junction, with 74 bp extending into the IRb region. The ndhF gene was 63 bp upstream of the SSC region, while ycf1 was located at the SSC/IRa junction, extending 107 bp into the IRa region. Additionally, the trnH gene was found at the IRa/LSC junction, with 16 bp located upstream in the LSC region.
These boundary differences suggest structural divergence at the junction sites between LSC, IR, and SSC regions, which may result from evolutionary rearrangements within the chloroplast genomes of T. sinensis from different geographical populations.
Phylogenetic analysis based on chloroplast protein-coding genes showed that our T. sinensis sequence was clustered with the T. sinensis sequence from GenBank (MN727386) with strong bootstrap support (90), and the two together formed a clade sister to Tinospora cordifolia. The phylogenetic tree also confirmed the monophyly of the Tinospora genus and clearly separated it from other related genera within Menispermaceae (Figure 3).
Phylogenetic tree of 29 Menispermaceae species inferred using the ML method based on 90 shared genes. Aconitum coreanum (MN400660.1) (Kim et al. 2019) and Aconitum vilmorinianum (OP227199.1) (unpublished) were selected as the outgroup. Numbers at the nodes represent the bootstrap values. The T. sinensis chloroplast genome obtained in this study (PV747743) is marked in bold. The sequences used for constructing the phylogenetic tree are as follows: Arcangelisia gusanlung (NC_058647.1) (Wen et al. 2021), Paratinospora sagittata (OQ579165.1) (unpublished), Fibraurea recisa (NC_060536.1) (Zheng and Feng 2022), T. cordifolia (NC_042153.1) (Monpara et al. 2024), Tinospora sinensis (MN727386.1) (direct submission), Sinomenium acutum (NC_046941.1) (Hina et al. 2020), Menispermum dauricum (OP271868.1) (direct submission), Menispermum canadense (OP271876.1) (direct submission), Pericampylus glaucus (NC_046846.1) (Kang and Wang 2019), Pachygone laurifolia (NC_072972) (direct submission), Eleutharrhena macrocarpa (NC_065487.1) (Song et al. 2022), Cyclea sutchuenensis (OR166993.1) (Wang et al. 2024), Cyclea hypoglauca (PQ369416.1) (direct submission), Stephania chingtungensis (OR166989.1) (Wang et al. 2024), Stephania longa (OR166982.1) (Wang et al. 2024), Stephania japonica (NC_029432.1) (Sun et al. 2016), Stephania dielsiana (NC_054337.1) (unpublished), Stephania herbacea (OR166991.1) (Wang et al. 2024), Stephania kwangsiensis (NC_048524) (Shi and Liu 2020), Stephania excentrica (OR166995.1) (Wang et al. 2024), Stephania dicentrinifera (OR166986.1) (Wang et al. 2024), Stephania kuinanensis (OR166996.1) (Wang et al. 2024), Stephania brachyandra (PQ399849.1) (Duong et al. 2025), Stephania yunnanensis (NC_087726.1) (direct submission), Stephania cephalantha (NC_067079.1) (Li et al. 2022), Stephania lincangensis (OR166998.1) (direct submission), Stephania epigaea (NC_058988.1) (direct submission), Stephania viridiflavens (OR166984.1) (Wang et al. 2024).
Discussion and conclusion
In this study, we assembled and annotated the complete chloroplast genome of T. sinensis, a medicinally important species within the family Menispermaceae. The chloroplast genome exhibits a typical conserved quadripartite structure.
Previous taxonomic records have referred to Tinospora sinensis (Lour.) Merr. as a synonym of T. cordifolia, reflecting historical ambiguities in species delimitation (Sharma et al. 2019). Morphological comparisons reveal that although T. sinensis and T. cordifolia share several traits, such as broadly ovate leaves, pubescence on the abaxial surface, and robust stems, they differ in stem surface characteristics, fruit morphology, and growth habits (Pandey et al. 2020).
Variation in the boundaries of IR/LSC and IR/SSC regions is frequently observed across chloroplast genomes and plays a crucial role in genome size variation. In this study, the observed differences in the location and extent of genes, such as rpl2, rps19, ndhF, ycf1, and trnH at the junctions suggest lineage-specific expansions or contractions of the IR regions in T. sinensis. These boundary shifts, although minor, may contribute to genomic rearrangements and evolutionary divergence among different populations or ecotypes of the same species.
In summary, this study provides the first comprehensive comparative analysis of the T. sinensis chloroplast genome, going beyond simple genome assembly to reveal key structural and evolutionary features. The identification of IR boundary variation involving rpl2 highlights potential lineage-specific structural divergence within T. sinensis, offering clues to chloroplast genome evolution in Menispermaceae. The detection of abundant SSRs and dispersed repeats provides valuable molecular markers for future studies on population genetics, species authentication, and resource conservation. Furthermore, phylogenetic reconstruction based on chloroplast protein-coding genes confirmed the monophyly of the Tinospora genus and resolved the placement of T. sinensis as a sister species to T. cordifolia with strong support. Collectively, these findings deepen our understanding of the genomic architecture and evolutionary relationships within Tinospora and establish a solid foundation for future taxonomic, phylogenomic, and medicinal studies.
Supplementary Material
Supplemental Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alves SIA et al. 2023. Easy SSR: a user-friendly web application with full command-line features for large-scale batch microsatellite mining and samples comparison. Front Genet. 14:122855237693309 10.3389/fgene.2023.1228552 PMC 10483286 · doi ↗ · pubmed ↗
- 2Amiryousefi A, Hyvönen J, Poczai P. 2018. I Rscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 34(17):3030–3031.29659705 10.1093/bioinformatics/bty 220 · doi ↗ · pubmed ↗
- 3Banerjee A et al. 2020. Protective efficacy of Tinospora sinensis against streptozotocin induced pancreatic islet cell injuries of diabetic rats and its correlation to its phytochemical profiles. J Ethnopharmacol. 248:112356.31669668 10.1016/j.jep.2019.112356 · doi ↗ · pubmed ↗
- 4Beier S et al. 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33(16):2583–2585.28398459 10.1093/bioinformatics/btx 198PMC 5870701 · doi ↗ · pubmed ↗
- 5Choudhuri S, Sau K. 2024. Codon U: a python package for codon usage analysis. IEEE/ACM Trans Comput Biol Bioinf. 21(1):36–44.10.1109/TCBB.2023.333582338015670 · doi ↗ · pubmed ↗
- 6Daniell H et al. 2016. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17(1):134.27339192 10.1186/s 13059-016-1004-2PMC 4918201 · doi ↗ · pubmed ↗
- 7Duong VT et al. 2025. The complete chloroplast genome sequence of the medicinal plant Stephania brachyandra Diels 1910 (Menispermaceae, Ranunculales) and phylogenetic analysis. Mitochondrial DNA Part B. 10(7):573–578.40524818 10.1080/23802359.2025.2519216 PMC 12168393 · doi ↗ · pubmed ↗
- 8Hina F et al. 2020. Characterization of the complete chloroplast genome of an important medicinal plant, Sinomenium acutum (Menispermaceae, Ranunculales). Mitochondrial DNA Part B. 5(2):1305–1306.