Peer review of the pesticide risk assessment of the active substance phenmedipham
Fernando Álvarez, Maria Arena, Domenica Auteri, Sofia Batista Leite, Marco Binaglia, Anna Federica Castoldi, Arianna Chiusolo, Angelo Colagiorgi, Mathilde Colas, Federica Crivellente, Chloe De Lentdecker, Isabella De Magistris, Mark Egsmose, Gabriella Fait, Franco Ferilli

TL;DR
This paper summarizes the peer review of phenmedipham, a pesticide, assessing its risks and regulatory compliance in the EU.
Contribution
The paper updates the risk assessment of phenmedipham with new evaluations on endocrine disruption and genotoxicity.
Findings
Phenmedipham's use as a herbicide on sugar beet/fodder beet was evaluated for regulatory compliance.
New concerns were identified regarding endocrine disrupting properties and genotoxicity.
Some required information for regulatory compliance is still missing.
Abstract
The conclusions of EFSA following the peer review of the initial risk assessments carried out by the competent authorities of the rapporteur Member State Finland and co‐rapporteur Member State Denmark for the pesticide active substance phenmedipham are reported. The context of the peer review was that required by Commission Implementing Regulation (EU) No 844/2012. The conclusions were reached on the basis of the evaluation of the representative uses of phenmedipham as a herbicide on sugar beet/fodder beet. The conclusions were updated with regard to the endocrine disrupting properties and the genotoxicity assessment following a mandate received from the European Commission in January 2019 and its update in January 2024, respectively. The reliable end points, appropriate for use in regulatory risk assessment, are presented. Missing information identified as being required by the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1 Figure 2
Figure 2 Figure 3
Figure 3 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6 Figure 7
Figure 7 Figure 8
Figure 8 Figure 9
Figure 9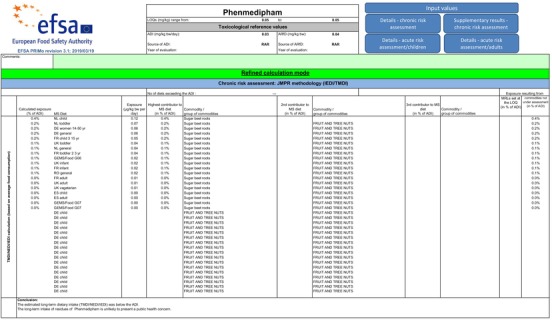 Figure 10
Figure 10 Figure 11
Figure 11 Figure 12
Figure 12| Line of evidence | Uncertainty | Comment |
|---|---|---|
|
| ||
| Study conducted to non‐standardised protocol | The only available study, extended amphibian metamorphosis assay (EAMA) is a non‐standardised protocol. No validation has been performed on this type of test and no positive control was included in the test; therefore, the performance of this test compared to standardised protocols is unknown. Nevertheless, the statistical power of the main apical T‐mediated parameter, time to reach stage NF 62, is comparable to other standardised test designs (Rizzuto et al., | The available EAMA has not been validated since it represents an enhancement of the existing standard AMA. |
| TPO inhibition positive in‐vitro | The ToxCast assay CCTE_Simmons_AUR_TPO_dn was positive although a flag was present for the purity of the substance. |
Available ToxCast information indicated that phenmedipham is positive in the AUR_TPO assay (CCTE_Simmons_AUR_TPO_dn) at a much lower concentration (0.31 μM) than the positive finding in the non‐specific protein inhibition observed in Quantilum_inhib_2_dn assay (9.5 μM). The potency values between the 2 assays are separated by approximately 1 log10‐μM, indicating that the TPO inhibition could be a specific hit and there is no strong evidence of interference with the non‐specific protein inhibition. The flag related to the purity of the substance is not something questioning the fact the assay is positive but only the endpoint setting. |
| Evidence of systemic toxicity | In the available EAMA 3 tadpoles died at day 3, 6 and 8 at the highest tested concentration leading to an overall 4% mortality at that concentration. However, no other signs of systemic toxicity were noted. | The mortality observed at the highest tested concentration, in absence of other signs of systemic toxicity, was more attributed to a sort of acute effects since after day 8, neither other mortalities were recorded nor other signs of systemic toxicity. Therefore, that concentration is not representing the MTC (maximum tolerated concentration). |
| Evidence of T‐mediated endocrine activity in the EAMA | Clear changes in the thyroid histology were noted in the available EAMA. It is not known if those findings were accompanied by changes in the thyroid hormone levels since thyroid hormone levels are not measured in the available amphibian study. | Changes in the thyroid histology are used as a proxy of the changes in the thyroid hormones. |
| Evidence of T‐mediated adversity in the available EAMA | A statistically significant delay in the time to reach NF62 was observed in the EAMA. | Although the relevance at the level of population of 1‐day delay in the time to reach NF62 may be questioned, no other evidence was submitted to demonstrate that the effect was not relevant, in line with Regulation 2018/605 stating the following: |
| Biological plausible link between the endocrine activity and the adversity | A biological plausible link between the endocrine activity (TPO inhibition and changes in thyroid histology) and T‐mediated adversity (delay in time to reach metamorphosis) was established by the coherence analysis, i.e. the underlying knowledge of the likely endocrine nature of the effects may be such that judgement can be reached on the biological plausibility of a link without a detailed MoA analysis. | Although a detailed MoA analysis was not available, the knowledge on the biologically plausible link of the observed endocrine activity and the T‐mediated adversity is well established by available literature (Haigis et al., |
| Criteria not met for mammals and humans | Potential difference in physiology and sensitivity between mammals and non‐mammalian species, leading to a different outcome of the ED assessment, could be expected, although they could not be clearly ruled out. Metabolism studies to explain different responses between vertebrate species are not available. |
The data set was considered sufficiently investigated and overall negative for humans. In the available Extended one generation study with rats at low doses, statistically significant decrease in T3 and T4 levels, observed only in the F1 generation cohort 1a, cannot be dismissed as being potentially related to a perturbation of the HPT axis after exposure to phenmedipham. However, these effects are not accompanied by a concomitant increase in TSH levels and changes in thyroid histology. Moreover, it should be emphasised that it could be expected to have different outcomes of the ED assessment for humans and non‐target organisms, e.g. methoxychlor (Pickford, |
|
| ||
| Positive evidence of EAS‐mediated endocrine activity in the FSTRA | Changes in the gonad histopathology in fish females accompanied by a decrease in fecundity were observed in absence of systemic toxicity. | A reduction in fecundity was observed at the highest tested concentration, which was accompanied by a reduction in POF. The reduction in POF is considered a consequence of the reduced fecundity and not a clear indication of an ED‐mediated effect. |
| Equivocal results for the in vitro H295R steroidogenesis assay (OECD TG 456) | The in vitro H295R steroidogenesis assay (OECD TG 456) provided equivocal evidence indicative of 17β‐oestradiol (E2) synthesis induction, a third confirmatory run was not conducted although it could be necessary. | No other positive in vitro findings were available. Although the in‐vitro studies are done using mammalian cells, they may provide useful mechanistic information, also considering that it is well‐known that the endocrine system is well‐conserved across vertebrates. |
| Criteria not met for mammals and humans | Potential difference in physiology and sensitivity between mammals and non‐mammalian species. Metabolism studies to explain different responses between vertebrate species are not available. | Although there may be some physiological differences, it is also well‐known that the endocrine system is well‐conserved across vertebrates. |
| Compound (name and/or code) | Persistence | Ecotoxicology |
|---|---|---|
|
| Low to high (DT50 = 4.2–139.5 days) | Low risk to in‐soil communities |
|
| Low to moderate (DT50 = 8.0–22.2 days) | Low risk to in‐soil communities |
| Compound (name and/or code) | Mobility in soil | > 0.1 μg/L at 1 m depth for the representative uses | Pesticidal activity | Toxicological relevance |
|---|---|---|---|---|
|
| Low ( | FOCUS GW: No | Yes | Yes |
|
| High to very high | FOCUS GW: No | Assessment not triggered for the representative uses evaluated | Assessment not triggered for the representative uses evaluated |
| Compound (name and/or code) | Ecotoxicology |
|---|---|
|
|
High chronic risk to aquatic invertebrates for the use pattern with 2 and 3 applications. Low risk to aquatic organisms for the use pattern that includes 1 application. Risk assessment cannot be finalised due to missing data on algae. |
|
| High chronic risk to aquatic invertebrates other than sediment organisms. |
|
| Low acute and chronic risk to aquatic organisms with mitigation measures. |
| Compound (name and/or code) | Toxicology |
|---|---|
|
| > 7.0 mg/L air/4 h (nose only) – no classification required |
| Representative use | Sugar beet/fodder beet, max 320 g a.s./ha | Sugar beet/fodder beet, max 2 × 320 g a.s./ha | Sugar beet/fodder beet, max 3 × 320 g a.s./ha | |
|---|---|---|---|---|
|
| Risk identified | |||
| Assessment not finalised | ||||
|
| Risk identified | |||
| Assessment not finalised | ||||
|
| Risk identified | |||
| Assessment not finalised | ||||
|
| Risk identified | |||
| Assessment not finalised | X1,2 | X1,2 | X1,2 | |
|
| Risk identified | X | ||
| Assessment not finalised | ||||
|
| Risk identified | |||
| Assessment not finalised | ||||
|
| Risk identified | 1/4 FOCUS scenarios | 2/4 FOCUS scenarios | |
| Assessment not finalised | X3 | X3 | X3 | |
|
| Legal parametric value breached | |||
| Assessment not finalised | ||||
|
| Legal parametric value breached | |||
| Parametric value of 10 μg/L | ||||
| Assessment not finalised | ||||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAgricultural safety and regulations · Pesticide Residue Analysis and Safety · Effects and risks of endocrine disrupting chemicals
SUMMARY
Commission Implementing Regulation (EU) No 844/2012 (hereinafter referred to as ‘the Regulation’) lays down the procedure for the renewal of the approval of active substances submitted under Article 14 of Regulation (EC) No 1107/2009. The list of those substances is established in Commission Implementing Regulation (EU) No 686/2012. Phenmedipham is one of the active substances listed in Regulation (EU) No 686/2012.
In accordance with Article 1 of the Regulation, the rapporteur Member State (RMS), Finland, and co‐rapporteur Member State (co‐RMS), Denmark, received an application from the Task Force on Phenmedipham, comprising of UPL Europe Ltd. and Bayer CropScience AG, for the renewal of approval of the active substance phenmedipham. Complying with Article 8 of the Regulation, the RMS checked the completeness of the dossier and informed the applicants, the co‐RMS (Denmark), the European Commission and the European Food Safety Authority (EFSA) about the admissibility.
The RMS provided its initial evaluation of the dossier on phenmedipham in the renewal assessment report (RAR), which was received by EFSA on 21 December 2016. In accordance with Article 12 of the Regulation, EFSA distributed the RAR to the Member States and the applicants of the Task Force on Phenmedipham, for comments on 20 February 2017. EFSA also provided comments. In addition, EFSA conducted a public consultation on the RAR. EFSA collated and forwarded all comments received to the European Commission on 27 April 2017.
Following consideration of the comments received on the RAR, it was concluded that additional information should be requested from the applicants, and that EFSA should conduct an expert consultation in the areas of mammalian toxicology, residues, environmental fate and behaviour, and ecotoxicology.
In accordance with Article 13(1) of the Regulation, EFSA should adopt a conclusion on whether phenmedipham can be expected to meet the approval criteria provided for in Article 4 of Regulation (EC) No 1107/2009 of the European Parliament and of the Council.
The conclusions laid down in this report were reached on the basis of the evaluation of the representative uses of phenmedipham as a post‐emergence herbicide on sugar beet/fodder beet, as proposed by the applicants. EFSA published its conclusion on the peer review of the pesticide risk assessment of phenmedipham on 31 January 2018. Subsequently, the conclusions were updated with regard to the endocrine disrupting properties and the genotoxicity assessment following a mandate received from the European Commission in January 2019 and its update in January 2024, respectively. Full details of the representative uses can be found in Appendix A of this report.
The uses of phenmedipham according to the representative uses proposed at the European Union (EU) level result in a sufficient herbicidal efficacy against the target weeds.
A data gap was identified for a search of the scientific peer‐reviewed open literature on the active substance and its metabolites relevant to consumer exposure.
In the area of identity, physical/chemical properties and analytical methods, data gaps were identified for spectra of the relevant impurities, for the content of the relevant impurities before and after storage and for a method for determination of the relevant impurities in the representative formulation.
In the mammalian toxicology area, data gaps were identified in relation to toxicokinetics, skin sensitisation, possible phototoxicity within UVB wavelength, the need for genotoxicity and/or repeat‐dose toxicity data on the plant and livestock metabolites, and data to address the toxicological relevance of most impurities present in the technical specifications. In addition, information on analytical methods used in the toxicological studies as well as acute toxicity studies on the representative plant protection product or equivalent formulation are missing. A critical area of concern is identified as the technical specifications proposed are not covered by the batches used in key toxicological studies.
In the residue section, several data gaps were identified related to the representative uses, including incomplete residue trials and processing studies, the lack of investigation of transfer of residues into animal commodities and missing toxicological data for several metabolites relevant for processed and animal commodities. Due to all identified data gaps, the consumer risk assessment via dietary intake could not be finalised.
With respect to the fate and behaviour in the environment, the necessary information was available to conduct the exposure assessment for groundwater and the environment. The applicant did not provide appropriate information to address the effect of water treatment processes on the nature of the residues that might be present in surface water, when surface water is abstracted for the production of drinking water. This has led to the identification of a data gap and results in the consumer risk assessment not being finalised.
A number of data gaps were identified in the field of ecotoxicology in relation to the risk to the algae and aquatic invertebrates and the risk to bees. The risk assessment to aquatic organisms (algae) could not be finalised.
It was concluded that phenmedipham does not meet the endocrine disruption (ED) criteria for humans as laid down in point 3.6.5 of Annex II to Regulation (EC) No 1107/2009, as amended by Commission Regulation (EU) 2018/605. For wild mammals as non‐target organisms, it can be concluded that phenmedipham does not meet the ED criteria for EATS‐modalities. Phenmedipham does not meet the criteria for non‐target organisms other than mammals through the EAS‐modalities; however, for the T‐modality for non‐target organisms other than mammals, it is concluded that phenmedipham meets the ED criteria as laid down in point 3.8.2. of Annex II to Regulation (EC) No 1107/2009, as amended by Commission Regulation (EU) 2018/605 (critical area of concern).
BACKGROUND
Commission Implementing Regulation (EU) No 844/20121 (hereinafter referred to as ‘the Regulation’) lays down the provisions for the procedure of the renewal of the approval of active substances, submitted under Article 14 of Regulation (EC) No 1107/2009.2 This regulates for the European Food Safety Authority (EFSA) the procedure for organising the consultation of Member States, the applicant(s) and the public on the initial evaluation provided by the rapporteur Member State (RMS) and/or co‐rapporteur Member State (co‐RMS) in the renewal assessment report (RAR), and the organisation of an expert consultation where appropriate.
In accordance with Article 13 of the Regulation, unless formally informed by the European Commission that a conclusion is not necessary, EFSA is required to adopt a conclusion on whether the active substance can be expected to meet the approval criteria provided for in Article 4 of Regulation (EC) No 1107/2009 within 5 months from the end of the period provided for the submission of written comments, subject to an extension of an additional 3 months where additional information is required to be submitted by the applicant(s) in accordance with Article 13(3).
In accordance with Article 1 of the Regulation, the RMS Finland and co‐RMS Denmark received an application from the Task Force on Phenmedipham, comprising of UPL Europe Ltd. and Bayer CropScience AG, for the renewal of approval of the active substance phenmedipham. Complying with Article 8 of the Regulation, the RMS checked the completeness of the dossier and informed the applicants, the co‐RMS (Denmark), the European Commission and EFSA about the admissibility.
The RMS provided its initial evaluation of the dossier on phenmedipham in the RAR, which was received by EFSA on 21 December 2016 (Finland, 2016).
In accordance with Article 12 of the Regulation, EFSA distributed the RAR to the Member States and the applicants of the Task Force on Phenmedipham, for consultation and comments on 20 February 2017. EFSA also provided comments. In addition, EFSA conducted a public consultation on the RAR. EFSA collated and forwarded all comments received to the European Commission on 27 April 2017. At the same time, the collated comments were forwarded to the RMS for compilation and evaluation in the format of a reporting table. The applicants were invited to respond to the comments in column 3 of the reporting table. The comments and the applicants' response were evaluated by the RMS in column 3.
The need for expert consultation and the necessity for additional information to be submitted by the applicants in accordance with Article 13(3) of the Regulation were considered in a telephone conference between EFSA, the RMS and co‐RMS on 20 June 2017. On the basis of the comments received, the applicants' response to the comments and the RMS's evaluation thereof, it was concluded that additional information should be requested from the applicants, and that EFSA should conduct an expert consultation in the areas of mammalian toxicology, residues, environmental fate and behaviour, and ecotoxicology.
The outcome of the telephone conference, together with EFSA's further consideration of the comments, is reflected in the conclusions set out in column 4 of the reporting table. All points that were identified as unresolved at the end of the comment evaluation phase and which required further consideration, including those issues to be considered in an expert consultation, were compiled by EFSA in the format of an evaluation table.
The conclusions arising from the consideration by EFSA, and as appropriate by the RMS, of the points identified in the evaluation table, together with the outcome of the expert consultation and the written consultation on the assessment of additional information, where these took place, were reported in the final column of the evaluation table.
A final consultation on the conclusions arising from the peer review of the risk assessment took place with Member States via a written procedure in December 2017, leading to the finalisation of the EFSA Conclusion (EFSA, 2018).
Commission Regulation (EU) 2018/6053 introduced new scientific criteria for the determination of endocrine disrupting (ED) properties, applicable as of 10 November 2018 to all applications for the approval/renewal of active substances, including pending applications. The peer review on the active substance phenmedipham was already completed at the time of entry into force of the new criteria, and an assessment of the ED potential in line with the ECHA/EFSA guidance (2018) document4 for this substance was not available.
Since on the basis of the EFSA Conclusion published on 31 January 2018, it was not possible for risk managers to conclude whether or not the active substance phenmedipham is an endocrine disruptor, on 14 January 2019, the European Commission requested EFSA to re‐assess the information and update its Conclusion on the ED potential of the substance in accordance with the new criteria in accordance with the provisions of Article 14(1a) of Commission Regulation (EU) No 844/2012, as amended by Commission Regulation (EU) 2018/1659. For this purpose, EFSA has performed an assessment of the ED properties of the active substance phenmedipham in line with the EFSA/ECHA (2018) guidance for further consideration in the peer review (EFSA, 2019), and distributed it to the Member States and the applicant (i.e. Task Force on Phenmedipham comprising of UPL Europe Ltd. and Bayer CropScience AG), for consultation and comments on 28 May 2019.
Following a consultation with Member States in the Pesticide Peer Review Experts' Meeting meeting 10 Mammalian toxicology – Ecotoxicology joint session (July 2019), it was concluded that phenmedipham does not meet the ED criteria for humans for the thyroid (T) modality according to point 3.6.5 of Annex II of Regulation (EC) No 1107/2009, as amended by Commission Regulation (EU) No 2018/605. However, additional testing was required to complete the data package for the oestrogen, androgen and steroidogenesis (EAS)‐modalities in relation to human health and to further investigate the ED properties of the substance for non‐target organisms. Therefore, in accordance with the provisions of Commission Regulation (EU) No 2018/1659,5 on 9 August 2019, the applicant was given the opportunity to submit, within a period of 30 months, additional information to address the approval criteria set out in points 3.6.5 and 3.8.2 of Annex II to Regulation (EC) No 1107/2009, as amended by Commission Regulation (EU) No 2018/605, and/or documentary evidence demonstrating that phenmedipham may be used such that exposure is negligible, and/or the conditions for application of the derogation under Article 4(7) of Regulation (EC) No 1107/2009 are met. The additional information submitted by the applicant on 27 January 2022 was subsequently evaluated by the RMS. EFSA received the updated RAR with the revised ED assessment from the RMS on 10 May 2022 (Finland, 2022).
A consultation on the revised RAR on the ED assessment made available by the RMS after the 30‐month clock stop was conducted with Member States, the applicant, EFSA and the public in June–August 2022. All comments received were collated in the format of a reporting table and were considered during the finalisation of the peer review. In addition, as a result of the consultation, in light of the comments received, expert's consultations with Member States at the Pesticides Peer Review Experts' TC 100 (mammalian toxicology) and 104 (ecotoxicology) were conducted in April 2023.
In January 2024, in the light of the latest available status of knowledge and risk assessment methodologies in relation to the genotoxicity assessment of phenmedipham, i.e. availability of the ECHA Risk Assessment Committee Opinion (ECHA RAC, 2019) and the EFSA Scientific Committee Opinion to assess the lines of evidence of bone marrow exposure (EFSA Scientific Committee;, 2017), the European Commission requested via a specific mandate the RMS to update the RAR to also include a revised genotoxicity assessment and, depending on the outcome, to derive toxicological reference values6 and carry out the dietary and non‐dietary risk assessment. In turn, EFSA was requested to peer review the updated risk assessment provided by the RMS and issue its updated conclusions also on these aspects in addition to the ED assessment. EFSA received the updated RAR with the requested assessment from the RMS on 12 February 2024. A round of consultations with Member States in the Pesticide Peer Review Experts' Meeting TC 163 (ED) and 164 (mammalian toxicology) took place in March 2025.
A final consultation on the updated conclusions arising from the peer review following the mandate from the European Commission took place with Member States via a written procedure in August–September 2025.
This conclusion report summarises the outcome of the peer review of the risk assessment of the active substance and the representative formulation, evaluated on the basis of the representative uses of phenmedipham as a post‐emergence herbicide on sugar beet/fodder beet, as proposed by the applicants. In addition, the conclusions were updated with regard to the endocrine disrupting properties and the genotoxicity assessment of phenmedipham following the mandate received from the European Commission on 14 January 2019 and its update on 16 January 2024. A list of the relevant end points for the active substance and the formulation for representative uses is provided in Appendix A.
In addition, a key supporting document to this updated conclusion is the peer review report (EFSA, 2017 updated in September 2025), which is a compilation of the documentation developed to evaluate and address all issues raised in the peer review, from the initial commenting phase to the conclusion. The peer review report comprises the following documents, in which all views expressed during the course of the peer review, including minority views, where applicable, can be found:
- the comments received on the RAR;
- the reporting table (26 June 2017 December 2022);7
- the evaluation table (19 December 2017, updated in September 2025);
- the reports of the scientific consultation with Member State experts (where relevant);
- the comments received on the assessment of the additional information (where relevant);
- the comments received on the EFSA endocrine disruption (ED) assessment (May 2019);8
- the comments received on the draft EFSA conclusion.
Given the importance of the RAR, including its revisions (Finland, 2017, 2022, 2025), and the Peer Review Report and the EFSA ED assessment (EFSA, 2019), all these documents are considered as background documents to this conclusion and thus are made publicly available.
It is recommended that this conclusion report and its background documents would not be accepted to support any registration outside the EU for which the applicant has not demonstrated that it has regulatory access to the information on which this conclusion report is based.
THE ACTIVE SUBSTANCE AND THE FORMULATION FOR REPRESENTATIVE USES
Phenmedipham is the ISO common name for 3‐[(methoxyformyl)amino]phenyl (3‐methylphenyl)carbamate (IUPAC).
The formulation for representative uses for the evaluation was ‘PMP SE160’, a suspoemulsion (SE) containing 160 g/L phenmedipham.
The representative uses evaluated were broadcast spray applications in the post emergence stage of beets (sugar and fodder), to control broadleaved weeds and grasses. Full details of the good agricultural practices (GAPs) can be found in the list of end points in Appendix A.
Data were submitted to conclude that the uses of phenmedipham according to the representative uses proposed at the EU level result in a sufficient herbicidal efficacy against the target weeds, following the guidance document SANCO/2012/11251‐rev. 4 (European Commission, 2014).
A data gap has been identified for a search of the scientific peer‐reviewed open literature on the active substance and its metabolites relevant to consumer exposure, dealing with side effects on health, and published within the 10 years before the date of submission of the dossier, to be conducted and reported in accordance with EFSA guidance on the submission of scientific peer‐reviewed open literature for the approval of pesticide active substances under Regulation (EC) No 1107/2009 (EFSA, 2011).
CONCLUSIONS OF THE EVALUATION
IDENTITY, PHYSICAL/CHEMICAL/TECHNICAL PROPERTIES AND METHODS OF ANALYSIS
1
The following guidance documents were followed in the production of this conclusion: SANCO/3029/99‐rev. 4 (European Commission, 2000a), SANCO/3030/99‐rev. 4 (European Commission, 2000b) and SANCO/825/00‐rev. 8.1 (European Commission, 2010).
The proposed specifications were supported by batch data from industrial scale productions and quality control (QC) data. The proposed minimum purity of the technical material is 970 g/kg. Toluene with maximum content of 2 g/kg, 3‐methylaniline and 3‐aminophenol with a maximum content of 1 g/kg each are considered relevant impurities. It should be noted that the relevance of other impurities is not concluded (see Section 2). The manufactured technical material meets the requirements of the existing FAO specification (AGP: CP/90, 1980) in terms of minimum purity; relevant impurities are not mentioned in the FAO specification.
The batches used in the (eco) toxicological assessment do not support the proposed new specification (for both sources) neither the original reference specification (from Bayer). This constitutes a critical area of concern for Sections 2 and 5.
The assessment of the data package revealed no issues that need to be included as critical areas of concern with respect to the identity, physical, chemical and technical properties of phenmedipham or the representative formulation; however, data gaps were identified for spectra of the relevant impurities and for the content of the relevant impurities before and after storage. The main data regarding the identity of phenmedipham and its physical and chemical properties are given in Appendix A.
Adequate methods are available for the generation of pre‐approval data required for the risk assessment. Methods of analysis are available for the determination of the active substance in the technical material and in the representative formulation and for the determination of the respective impurities in the technical material. However, a data gap was identified for a validated method for determination of the relevant impurities in the representative formulation.
Phenmedipham residue can be monitored in food and feed of plant origin by the DFG method S19 (extended revision) using liquid chromatography with tandem mass spectrometry (LC–MS/MS) with a limit of quantification (LOQ) of 0.01 mg/kg in each commodity group. In addition, there is the QuEChERS method using gas chromatography–mass spectrometry (GC–MS) and/or LC–MS/MS for all plant commodities with LOQs in the range 0.01–0.05 mg/kg. It should be noted that a residue definition for monitoring in plant processed commodities is proposed. In case a specific maximum residue level (MRL) for these commodities is set, monitoring methods for the components included in the residue definition might be required. Residues of MHPC in food of animal origin can be determined by the QuEChERS method using LC–MS/MS with LOQ of 0.01 mg/kg in all animal matrices.
Residues of phenmedipham and MHPC in soil can be monitored by the DFG method S19 (extended revision) with LC–MS/MS with a LOQ 0.01 mg/kg.
An appropriate LC–MS/MS method exists for monitoring of phenmedipham and MHPC residues in water with a LOQ of 0.05 μg/L. Phenmedipham residues in air can be monitored by RP/HPLC‐UV with a LOQ of 10 μg/m^3^.
The LC–MS/MS method with a LOQ of 50 μg/L can be used for monitoring of phenmedipham and MHPC in body fluids. The method for monitoring in animal products can be used for determination of phenmedipham and MHPC in body tissues.
MAMMALIAN TOXICITY
2
The toxicological profile of the active substance phenmedipham was discussed at the Pesticides Peer Review Experts' Meetings 168 (October 2017), in TC 100 (April 2023) and in TC 164 (March 2025) and assessed based on the following guidance documents: SANCO/221/2000‐rev. 10‐final (European Commission, 2003), SANCO/10597/2003‐rev. 10.1 (European Commission, 2012), Guidance on dermal absorption (EFSA PPR Panel, 2012), EFSA Scientific Committee (2017).
The technical specifications proposed by both applicants, including the original one from Bayer are not representative of the batches tested in the toxicological studies, leading to a critical area of concern (see Section 10.2). In addition, the assessment of the toxicological relevance of most impurities has not been addressed (data gap). Toluene, 3‐aminophenol and 3‐methylaniline are relevant impurities due to their hazard but their maximum levels proposed for the technical specifications (2 g/kg, 1 g/kg and 1 g/kg, respectively) is not of toxicological concern. Analytical methods for rodent diet have been provided in the respective section of the RAR; it has to be further clarified which method has been used in each of the toxicological studies (data gap).
Phenmedipham absorption is relatively fast and extensive in the low doses (about 80% in 24 hours). Phenmedipham is widely distributed with higher amounts in plasma, whole blood, lungs, ovaries, thyroid gland, skin, pituitary, heart, adrenal glands, kidneys, spleen and liver. Around 90% of phenmedipham is excreted within 24 hours through the urine after low‐dose administration, while at high doses the elimination is less fast and mainly through faeces. Basic toxicokinetic data have not been investigated in rodents and a data gap was identified. Phenmedipham is extensively metabolised in the rat via oxidative/hydrolytic cleavage followed by hydroxylation, acetylation and oxidation reactions. The major component in faeces is the parent compound. In the comparative interspecies (rat and human) metabolism study in vitro, significant differences and human specific metabolites were not observed. Since the metabolite MHPC has been identified as a major metabolite in rats, it should be included in the residue definition for monitoring in body fluids (blood, plasma and urine) in humans together with the parent phenmedipham.
Low acute toxicity was observed when phenmedipham was administered by the oral, dermal or inhalation routes; no skin or eye irritation was attributed to the active substance. A conclusion regarding the potential for phenmedipham to cause skin sensitisation cannot be drawn due to insufficient data (data gap). Phenmedipham did not show phototoxic potential in the OECD 3T3 NRU‐PT test. The OECD 3T3 NRU‐PT test might not be an appropriate test for UVB absorbers such as phenmedipham. However, no validated methods are available to address properly UVB absorbers (data gap).
In all short‐term studies in rats, mice and dogs, the critical effects observed were related to haemolytic anaemia (increased methaemoglobin (MetHb), decrease in haemoglobin, haematocrit and red blood cells, increased extramedullary haematopoiesis and hemosiderin deposition in spleen, liver and kidneys). The overall short‐term no‐observed adverse effect level (NOAEL) is 3.5 mg/kg body weight (bw) per day from the 90‐day rat studies.
Phenmedipham did not induce gene mutation in vitro, but it is clastogenic in vitro as demonstrated by the positive in vitro chromosomal aberration assays in human lymphocytes and in Chinese hamster ovary cells. The in vitro test battery did not address properly aneugenicity since an in vitro micronucleus (MN) test was not available. In vivo mouse MN assay results were negative, and data from quantitative whole‐body autoradiography and plasma analysis demonstrated that the bone marrow was reached. However, during the Pesticide Peer Review meeting TC 164,9 Member State (MS) experts could not reach a consensus on whether the bone marrow had been exposed sufficiently.10 EFSA considered that the lines of evidence in the available toxicological data set, analysed using a weight of evidence (WoE) approach in accordance with EFSA Scientific Committee, 2017, are sufficient to demonstrate the bone marrow exposure. Overall, EFSA considers phenmedipham unlikely to be genotoxic in vivo. Some experts agreed with EFSA, while the slight majority of experts, including the RMS, disagreed and supported the position of an insufficient exposure of bone marrow that would lead to inconclusive outcome of the genotoxicity assessment in the presence of positive results for in vitro clastogenicity which was not possible to be excluded in in vivo studies and to data gap for aneugenicity.
Germ cell mutagenicity hazard class was not discussed by the ECHA Risk Assessment Committee in 2019 (ECHA RAC, 2019), and genotoxicity was presented only as background information for carcinogenicity assessment in the CLH report.11 The ECHA RAC, in line with the dossier submitter (Finland), did not consider the available data to raise a significant concern about genotoxicity.
The relevant long‐term NOAEL is 3 mg/kg bw per day from a 2‐year study in rats, based on haemolytic anaemia (hemosiderin deposition in spleen and liver) as well as histopathological effects in pituitary and kidney. An increased incidence of endometrial stromal sarcoma and of pituitary adenoma was observed in the 2‐year studies in rats. The ECHA RAC concluded that no classification for carcinogenicity is warranted for phenmedipham (ECHA RAC, 2019) and such proposal has been legally implemented (ATP 17).12
Two main and one supplementary multigenerational reproductive studies in rats were submitted for phenmedipham. An extended one‐generation reproductive toxicity (EOGRT) study in rat is also available for phenmedipham; this study is considered relevant for the derivation of reproductive, parental and offspring NOAELs.13The reproductive NOAEL is 200 mg/kg bw per day (the highest dose tested). The parental NOAEL is 15 mg/kg bw per day based on hemosiderin pigment deposition in kidney and spleen of F0 males and on haematological findings observed starting from 60 mg/kg bw per day in F0 males. The NOAEL for offspring is 15 mg/kg bw per day based on hemosiderin pigment deposition in kidney in F1 males and spleen in F1 females and haematological findings observed starting from 60 mg/kg bw per day in F1 males.
Three acceptable developmental studies were provided (one in rat and two in rabbits). For both maternal and developmental toxicity in rats a lowest observable adverse effect level (LOAEL) is set at the low dose of 150 mg/kg bw per day based on reduced body weight gain in dams and occurrence of runts in all doses. For rabbits, a maternal NOAEL is set at 225 mg/kg bw per day based on reduced body weight gain and reduced food consumption. The developmental NOAEL was also set at 225 mg/kg bw per day based on reduced body weight and retarded ossification. The ECHA RAC concluded that no classification for reproductive and/or developmental toxicity is warranted for phenmedipham (ECHA RAC, 2019) and such proposal has been legally implemented.
Overall, the available data for phenmedipham do not raise concern in relation to neurotoxicity or immunotoxicity.
During the first peer review of phenmedipham (European Commission, 2004), an acceptable daily intake (ADI) of 0.03 mg/kg bw per day was derived, on the basis of the 2‐year rat study and applying an uncertainty factor of 100. An acute reference dose (ARfD) was not allocated, and an acceptable operator exposure level (AOEL) of 0.13 mg/kg bw per day was established on the basis of the 90‐day rat study (uncertainty factor (UF) 100, no correction for oral absorption). The previous established ADI is confirmed. The ARfD is 0.04 mg/kg bw, based on the 2‐year rat study (based on increased methaemoglobin levels observed in week 13) applying an increased UF of 200 to extrapolate from a LOAEL to NOAEL.14 The same value is proposed for the acute acceptable operator exposure level (AAOEL). The AOEL is 0.04 mg/kg bw per day based on 90‐day rat study,15 applying an uncertainty factor of 100 and no correction factor for oral absorption.
Acute toxicity studies were not submitted for the representative plant protection product (PPP) or equivalent formulation; some endpoints regarding the acute toxicity were determined by the use of component analysis approach and also a local lymph node assay (LLNA) study was submitted, however the toxicity of the plant protection product and its relation to the toxicity of the active substance, adverse effects and relative hazard associated with the different routes of exposure cannot be fully drawn (data gap). The dermal absorption values based on an in vitro human study using the representative formulation for phenmedipham are 0.2% for the neat formulation (160 g/L), 1% for the spray dilution (2 g/L), and 2.5% for pro‐rata correction (0.8 g/L).
EFSA provided calculations following the pertinent EFSA guidance documents (EFSA, 2014a, 2014b, 2022) (see details Appendix D), considering the representative uses on sugar beet/fodder beet and the agreed toxicological endpoints. For the operators, exposure estimates are below the (A)AOEL for all uses when the application is vehicle‐mounted (and not handheld or knapsack). For the workers, performing either reaching/picking, inspection or removal of bolting beet, the exposure estimates are below the AOEL for all uses. For the residents and bystanders, the estimated exposure is below the (A)AOEL and no specific risk mitigation measure is required for the supported uses (see Appendix D).
A number of metabolites were found in significant amounts in residues. MHPC has been identified as a major metabolite in the rat and the reference values of phenmedipham, are applicable to this metabolite. For 3‐methylaniline (m‐toluidine), which has harmonised classification as Acute tox 3 (H301, H311, H331: toxic if swallowed, if inhaled and in contact with skin), and STOT‐RE 2 (H373: may cause damage to organs through prolonged or repeated exposure), genotoxicity and repeated‐dose toxicity data relevant to consumer exposure are needed. Regarding metabolites 3‐aminophenol, 3‐acetamidophenol, 4‐acetamido‐o‐cresol, 4‐amino‐2‐methylphenol, acetamido‐benzoic acid, aminobenzoic acid and m‐acetotoluidine, their genotoxic potential has to be addressed, and pending on further assessment in the residue section, repeated‐dose toxicity data relevant to consumer exposure may be needed (data gap).
RESIDUES
3
The assessment in the residue section is based on the OECD guidance document on overview of the residue chemistry studies (OECD, 2009), the OECD publication on the MRL calculations (OECD, 2011) and the European Commission guideline document on the MRL setting (European Commission, 2011).
Phenmedipham was discussed at the Pesticides Peer Review Experts' Meeting 167 (October 2017).
Metabolism of phenmedipham in primary crops was investigated upon foliar application in roots/tuber crops (sugar beet) with both [amino‐phenyl‐UL‐^14^C] and [phenyl‐methyl‐UL‐^14^C] phenmedipham at max 1069 g/ha application rate, and in fruits (strawberries) only with [amino‐phenyl‐UL‐^14^C] radiolabelled phenmedipham at max 2880 g/ha application rate.
Phenmedipham and its conjugates were the predominant compounds of the total residues in sugar beet in immature and mature leaves (95% total radioactive residue (TRR) and 51% TRR, respectively). In sugar beet, root phenmedipham and its conjugates were detected at a low level (6.6% TRR) while a major unknown fraction accounted for ca. 26% TRR in roots and 14% TRR in maturity leaves. This fraction was generated only from the amino phenol moiety and constituted of several polar minor metabolite fractions. The metabolism data in sugar beet were considered sufficient to support the representative uses on sugar beet, except one MS who considered that a new metabolism study in root crops should be provided in view of the authorised uses on other root crops. In strawberries, phenmedipham was the main compound recovered in fruits (58% TRR) while 3‐acetamidophenol compound accounted for 13% TRR. 3‐acetamidophenol is a rat metabolite and it was not recovered in the sugar beet metabolism study. Based on these metabolism studies, the residue definition for enforcement was derived as phenmedipham restricted to roots and fruit crops only. For risk assessment residue definition, the experts were of the opinion, that in addition to phenmedipham also glucoside conjugates should be included and the residue definition should be restricted to sugar beet only. For strawberries, since only one label was investigated, no residue definition for risk assessment was proposed. Provisional conversion factors (CFs) for risk assessment in sugar beet of 1.4 (root) and 1.2 (leaves) were derived from the metabolism studies.
Under standard hydrolysis conditions when investigated with phenyl‐methyl labelling, phenmedipham degraded partially into 3‐methylaniline (m‐toluidine) at baking/brewing and boiling (86% applied radioactivity (AR)) and completely into 3‐methylaniline (m‐toluidine) under sterilisation conditions. Under these harsh conditions, it can reasonably be assumed that the formation of aniline can be excluded. For the amino phenol labelling form, a complete degradation of phenmedipham to MHPC metabolite was observed at baking/brewing and boiling and also in conditions representative of sugar production. Under pasteurisation conditions, phenmedipham is considered stable for both labelling forms (82%–87% of AR). The residue definitions for monitoring in processed commodities is sum of phenmedipham and MHPC, expressed as phenmedipham, while for risk assessment is separately 3‐methylaniline (m‐toluidine) (see data gap in Section 2) and sum of phenmedipham and MHPC. The possible formation of MHPC and 3‐methylaniline (m‐toluidine) in sugar and sugar beets by‐products, used as a feed item for animals, has to be also investigated (data gap).
A confined rotational crop study was conducted on wheat, turnip and chard with phenyl‐methyl‐UL‐^14^C phenmedipham at 30, 164 and 305 plant‐back intervals (PBIs). Although some deficiency was noted (low rate of identification), the metabolism pattern was considered sufficient because of the expected low residue levels in all edible parts. A second study was conducted with the amino‐phenyl‐UL‐^14^C phenmedipham form on lettuce, sugar beet and wheat at 30, 120 and 365 PBIs. The metabolic pattern was consistent throughout all PBIs with phenmedipham and MHPC (major soil metabolite) being the only identified metabolite in rotational crops. In wheat straw, phenmedipham (20% TRR) and MHPC (25% TRR) were the major compounds of the TRR (0.95 mg/kg). The same metabolic pattern was observed in cereal forage. A significant decline of the total residues from the first to the third rotation interval was observed. This is confirmed by the field rotational crop trials conducted on leafy (lettuce), root crops (carrots and turnip), and cereals (wheat and barley) showing that no residues of phenmedipham and MHPC are above 0.01 mg/kg. Based on the confined rotational studies, the risk assessment residue definition is proposed as sum of phenmedipham and MHPC, free and conjugates, expressed as phenmedipham, while for enforcement the residue definition is set as phenmedipham only. For cereal fodder commodities, an average CF of 1.7 (1.3 for forage and 2.1 for straw) was derived based on the rotational metabolism studies while for sugar beet the same CFs as for primary crops are applicable. However, these CFs should be regarded as provisional and have to be confirmed by the field residue trials.
Storage stability data demonstrated that phenmedipham and MHPC residues are stable up to 24 months in high water, high oil, high protein, high starch and high acid content commodities, when stored at –20°C.
The submitted residue trials from SEU are not valid since they were not compliant with the representative GAP. For northern Europe (NEU), three fully compliant and one slightly overdosed residue trials analysed according to the risk assessment residue definition were submitted for sugar beet roots, while for the leaves only phenmedipham was investigated. Since a no‐residue situation (lower than 0.01 mg/kg) cannot be confirmed, and no valid residue trials are available for southern Europe (SEU), sufficient number of residue trials for sugar beet roots and leaves compliant with the representative GAPs and analysed according to the risk assessment residue definition should be submitted (data gap).
Livestock metabolism studies were investigated with both methyl phenyl and amino labels in lactating goats for 3 days with a dose rate of 0.1 mg/kg bw per day. Although the dosing period and the storage of the samples for 8 months (milk) and 7 months (other tissues) is not compliant with the current recommendations, the studies were found acceptable to elucidate the metabolic pattern. Phenmedipham was extensively metabolised in ruminants resulting in a high number of compounds. The major compound were MHPC up to (38% TRRs) in milk (45% TRRs) in kidney and (34% TRRs) in liver, 3‐aminophenol (16% TRRs) in kidney and (37% TRRs) in liver, 3‐acetamidophenol in milk (23% TRRs) and (15% TRRs) in kidney, 4‐acetamido‐o‐cresol (47% TRRs), 22% TRRs in milk, 4‐amino‐2‐methylphenol (24% TRRs) in kidney and (28% TRRs) in liver. In addition, acetamido‐benzoic acid, aminobenzoic acid and m‐acetotoluidine were also present for more than 10% of TRRs in liver. In muscle and fat no metabolites identification was possible due to the very low TRRs recovered in these matrices (< 0.01 mg/kg). Preliminarily, taking into account the information available regarding the occurrence and toxicity of the recovered residue compounds, the residue definition for risk assessment for ruminants should include MHPC, 3‐aminophenol, 3‐acetamidophenol, 4‐acetamido‐o‐cresol, 4‐amino‐2‐methylphenol, acetamido‐benzoic acid, aminobenzoic acid and m‐acetotoluidine. For enforcement, the residue definition is proposed as MHPC expressed as phenmedipham.
As regards the feeding studies, they were not submitted although based on the available data, they are triggered for all animal diets, except poultry (data gap). The feeding studies have to be covered by the storage stability data and validated analytical methods.
The livestock residue assessment should be regarded as provisional, pending on the finalisation of the assessment on processed commodities, the submission of the additional GAP compliant trials in sugar beet roots and leaves, and the outcome of the toxicological evaluation of all compounds included in the residue definition (see Sections 2 and 9).
Fish metabolism studies were not triggered.
A preliminary consumer dietary risk assessment was conducted by EFSA (see Appendix D) for phenmedipham by using the ADI of 0.03 mg/kg bw per day, the ARfD of 0.04 mg/kg bw (see Section 2), an input values of 0.05 mg/kg for phenmedipham in sugar beet resulting from the limited number of valid trials (see data gap above in field trials) and the conversion factor of 1.4 for phenmedipham glucoside conjugates derived from the metabolism studies. The provisional dietary intake calculation resulted in a chronic exposure (theoretical maximum daily intake (TMDI)) of 0.4% of the ADI (NL diet), and acute exposure (international estimated short‐term intake (IESTI)) of 4% of the ARfD for sugar (processed commodity). It is highlighted that these calculations are provisional, and the consumer risk assessment is not finalised (see Section 10.1). This is due to the incomplete data set from residue trials in sugar beet roots and leaves, the lack of investigation of the transfer of residues into animal commodities and missing toxicological data for several metabolites included in residue definitions for risk assessment. These metabolites are 3‐aminophenol, 3‐acetamidophenol, 4‐acetamido‐o‐cresol, 4‐amino‐2‐methylphenol, aminobenzoic acid and m‐acetotoluidine (relevant for animal commodities) and 3‐methylaniline (relevant for processed commodities) (see Section 2).
The consumer risk assessment from the consumption of drinking water is not finalised considering the lack of appropriate information to address the effect of water treatment processes on the nature of residues, potentially present in surface water, when surface water is abstracted for the production of drinking water (see Section 4).
Since during the peer review for the renewal of phenmedipham approval different toxicological endpoints as well as residue definitions for risk assessment in plant and animal commodities were derived, the MRLs derived under Article 12 of the Regulation (EC) No 396/2005 should be revised (EFSA, 2014a, 2014b).
Considering that sugar beet and fodder beet are harvested before the flowering, and provided that they are not cultivated for seed production, it can be reasonably assumed that bees will usually not get in contact with pollen and therefore the determination of residues in pollen and bee products is not considered necessary.
ENVIRONMENTAL FATE AND BEHAVIOUR
4
Phenmedipham was discussed at the Pesticides Peer Review Experts' Teleconference 151 (5 October 2017).
The rates of dissipation and degradation in the environmental matrices investigated were estimated using FOCUS (2006) kinetics guidance. In soil laboratory incubations under aerobic conditions in the dark, phenmedipham exhibited low to high persistence, forming the metabolite MHPC (max 14% AR), which exhibited low to moderate persistence. Mineralisation of the ^14^C methyl‐aniline ring radiolabelled phenmedipham to carbon dioxide accounted for a maximum of 28.7% AR and the ^14^C‐amino‐phenol labelled phenmedipham for a maximum of 20.2% AR after 112 days. The formation of unextractable residues for the ^14^C‐aniline ring and the ^14^C‐amino‐phenol radiolabel accounted for maxima of 61.5% AR and 73.8% AR after 112 days, respectively. In the available anaerobic soil incubation, phenmedipham exhibited moderate persistence and produced the metabolite MHPC (max 45.3% AR). Photolysis could marginally contribute to the degradation of phenmedipham in soil.
Field dissipation studies with phenmedipham were available in Germany and USA (California) in the first peer review. These studies have been considered not to provide reliable degradation half‐lives during the renewal assessment.
Phenmedipham is expected to exhibit low mobility in soil and metabolite MHPC exhibited very high to high mobility on the basis of the available batch adsorption desorption studies.
In the available lysimeter studies (in the UK and Germany) of 2 or 3 years of duration, phenmedipham and MHPC were not found in the leachates at levels above 0.01 μg/L. Total radioactivity in the leachate amounted up to a maximum of 1.9 μg/L in the first year in the UK lysimeter study, attributed to humic acid type fragments and incorporated radioactivity in natural components.
At environmental relevant temperature, phenmedipham undergoes rapid aqueous hydrolysis under pH above 6 but slow or very slow hydrolysis at more acidic conditions. Photolysis in water is not expected to contribute to the degradation of phenmedipham in aquatic environment. In laboratory incubations in dark aerobic natural sediment water systems (pH 6.0–8.35), phenmedipham exhibited very low persistence, forming the major metabolites MHPC (max ca. 97.1% AR in whole system after 1 day, exhibiting low to medium persistence) and 3‐methylaniline (m‐toluidine) (max 77% in whole system, very low to medium persistent). The unextractable sediment fraction was the major sink for the methyl‐aniline and amino‐phenol rings ^14^C radiolabel moieties, accounting for up to maxima 73.4% AR and 78.0% AR, respectively, at the end of the study. Mineralisation accounted for 54.8% AR and 34.1% AR for the methyl‐aniline and amino‐phenol moieties at the end of the study. The necessary surface water and sediment exposure assessments (predicted environmental concentrations (PEC) calculations) were carried out for the metabolites MHPC and 3‐methylaniline (m‐toluidine) using the FOCUS (2001) step 1 and step 2 approach (version 2.1 of the Steps 1–2 in FOCUS calculator). For the active substances phenmedipham and 3‐methylaniline (m‐toluidine) also, step 3 (FOCUS, 2001) PEC SW/SED calculations were available. FOCUS Step 4 calculations considering different buffer zones in combination with mitigation by drift reducing nozzles were conducted based on the Step 3 results for phenmedipham and 3‐methylaniline. The step 4 calculations following the FOCUS (2007) guidance, with no‐spray drift buffer zones being together with drift reduction nozzles were implemented in the values used for the risk assessment (only values leading to a realistic spray drift mitigation up to 95% are reported in Appendix A). The SWAN tool (version 1.1.4) was used to implement these mitigation measures in the simulations.
The groundwater exposure assessments were carried out using FOCUS (2009) scenarios and the models PEARL v.4.4.4, PELMO v.5.5.3 and MACRO v.5.5.4 (Châteadun) for the active substance phenmedipham and metabolite MHPC. The potential for groundwater exposure from the representative uses by phenmedipham and MHPC above the parametric drinking water limit of 0.1 μg/L was concluded to be low in geoclimatic situations that are represented by all nine FOCUS groundwater scenarios.
The applicant did not provide appropriate information to address the effect of water treatment processes on the nature of the residues that might be present in surface water when surface water is abstracted for the production of drinking water. This has led to the identification of a data gap (see Section 7) and results in the consumer risk assessment not being finalised (see Section 9).
The PEC in soil, surface water, sediment and groundwater covering the representative uses assessed can be found in Appendix A of this conclusion.
ECOTOXICOLOGY
5
The risk assessment was based on the following documents: European Commission (2002a, 2002b), SETAC (2001), EFSA (2009), EFSA PPR Panel (2013) and EFSA (2013). According to Regulation (EU) No. 283/201316 data should be provided regarding the acute and chronic toxicity to honeybees and data to address the development of honeybee brood and larvae. As the European Commission (2002a) does not provide a risk assessment scheme which is able to use the chronic toxicity data for adult honeybees and the honeybee brood, when performing the risk assessment according to European Commission (2002a), the risk to adult honeybees from chronic toxicity and the risk to bee brood, could not be finalised due to the lack of a risk assessment scheme. Therefore, the EFSA (2013) was used for risk assessment in order to reach a conclusion for the representative uses.
Phenmedipham was discussed at the Pesticides Peer Review Experts' Meeting 169 (October 2017)17 and 104 (April 2023) regarding the ecotoxicological relevant endpoint for wild mammals.
It is considered that insufficient information was provided to demonstrate the compliance of the batches used in the (eco)toxicity studies compared to the technical specification and therefore this issue was identified as critical area of concern.
The first tier risk assessment to birds and mammals indicated a low acute risk from dietary exposure for all representative uses. The long‐term risk to birds was concluded low for representative use in sugar beet/fodder beet with the worst‐case use pattern of three applications at 320 g a.s./ha; therefore, it could be considered low for all representative uses. Following the submission of additional data in line with the provisions as laid down in Regulation 2018/1659, the endpoint provisionally set18 for conducting the long‐term risk assessment to mammals was confirmed. As consequence, low long‐term risk to mammals could be concluded for the representative uses consisting of one and two applications. To refine the risk for the representative use with three applications, residue decline studies were available. However, those were considered acceptable for the NEU but not for SEU. Therefore, high risk was still concluded for that use.
The risk from secondary poisoning was not triggered (log P ow <3) and the risk to birds and mammals via consumption of contaminated water was assessed as low.
For aquatic organisms, toxicity data with the active substance were available on fish, aquatic invertebrates including sediment dwelling organisms and aquatic macrophytes. Ecotoxicity data on algae is missing for the active substance (data gap). Being algae the most likely risk assessment‐driver in the aquatic system due to the herbicidal activity of phenmedipham, the risk assessment cannot be finalised.
With regard to the pertinent aquatic metabolites in the surface water and sediment compartment, several ecotoxicity data with MHPC and 3‐methylaniline (m‐toluidine) were available on aquatic organisms.
The acute risk assessment for fish, aquatic invertebrates, sediment dwelling organisms and aquatic macrophytes is low for all representative uses for phenmedipham.
The chronic risk assessment for the worst‐case representative use in sugar beet (3 × 320 g/ha) indicated that phenmedipham is of high risk for aquatic invertebrates in two of four FOCUS Step 4 exposure scenarios even considering mitigation options (no‐spray buffer zones and vegetated buffer strips up to 20 m) and also in one of four FOCUS step 4 scenarios considering mitigation options (no‐spray buffer zones and vegetated buffer strips up to 20 m) for the use pattern that includes two applications at 320 g/ha (data gap). No chronic risk has been identified for invertebrates following the use pattern of one application at 320 g/ha when using mitigation measures up to 20 m. To add‐on, the chronic risk to fish is low if mitigation measures are considered (no‐spray buffer zones and vegetated buffer strips up to 20 m) for the worst‐case use in sugar beet.
The risk assessment indicated that MHPC is of low risk to aquatic organisms except to aquatic invertebrates (chronic), where, for the worst‐case representative use in sugar beet followed by three applications at an application rate of 320 g a.s./ha, the risk assessment indicated high risk in a screening step. No risk assessment was performed for the use patterns that include one and two applications (data gap).
For the worst‐case representative use, the risk assessment from 3‐methylaniline (m‐toluidine) fails to aquatic invertebrates in two of four FOCUS Step 3 scenarios (acute and chronic). Low risk to aquatic invertebrates is confirmed by using mitigation option measures as follows: no‐spray buffer zones and vegetated buffer strips up to 20 m.
Acute contact and oral toxicity studies on honeybees were performed with the active substance and the formulated product. Furthermore, a 10‐day chronic laboratory study with a phenmedipham‐based formulated product was available. The available ecotoxicity study with bumblebees showed that the active substance is equally toxic for bumblebees as for honeybees in a contact acute scenario; however, oral toxicity data to bumblebees is not available. According to EFSA (2013), low risk has been identified to honeybees from contact exposure for all representative uses. High risk has been identified in the oral chronic scenario due to weeds and in the treated crop. However, since the use patterns are applicable at early growth stage of the crop, the chronic risk to bees in the treated crop can be considered low unless Member States granted authorisations for seed production; in that case, this risk should be further considered. Likewise, since phenmedipham is particularly used for the control of a wide range of broad‐leaved weeds, the exposure via contaminated weeds could be considered of low relevance for the uses according to the GAPs reported.
The acute and chronic risk through exposure via residues in guttation fluid and via surface water was assessed as low in pertinent lower tier risk assessments according to EFSA (2013). However, the risk to bees should be evaluated for the puddle scenario (data gap). For honeybee larvae, a tier 1 risk assessment was not available due to the lack of a suitable endpoint according to the EFSA (2013). An Oomen et al. (1992) feeding test was available and no effects were observed; however, these kinds of studies are considered not suitable for risk assessment according to the EFSA (2013) (data gap). Insufficient information was available to perform a risk assessment for sub‐lethal effects (i.e. hypopharyngeal glands (HPGs), data gap) and accumulative effects. The risk from exposure to metabolites occurring in pollen and nectar from the representative uses in sugar beet is considered low provided that the use is at early stage of the sugar beet and assuming no seed production. Data to perform a risk assessment for solitary bees were not available and for bumble bees only the acute contact exposure scenario has been confirmed to be of low risk.
As regards to other non‐target arthropods, laboratory studies were available with the standard indicator species and the formulated product. No additional test species were tested at tier 1 but at higher tiers. On the basis of a risk assessment with the standard tier 1 indicator species, a high in‐field risk to non‐target arthropods was indicated for the representative worst case use of 3 applications on sugar beet/fodder beet. No off‐field risk from phenmedipham uses has been identified. A number of higher tier studies (extended laboratory and aged residue studies) with different arthropod species were available. These studies confirmed that a high‐initial in‐field risk can be refined; therefore, low risk to non‐target arthropods was concluded according to the representative worst case use.
Effects on non‐target soil meso‐ and macrofauna (i.e. earthworms, collembolan and soil predatory mites) has been tested with the active substance, the formulated product, the metabolite MHPC and as additional information, the metabolite 3‐methylaniline (m‐toluidine) was tested (only one earthworms' test study). In the first tier, low risk has been identified for all in‐soil communities including soil microorganisms based on the representative worst case use of 3 applications at an application rate of 320 g a.s./ha.
Low risk was identified on non‐target terrestrial plants and for organisms involved in biological methods for sewage treatment.
ENDOCRINE DISRUPTION PROPERTIES
6
The assessment of the ED potential of phenmedipham was initially discussed at the Pesticides Peer Review Experts' meeting PREV 10 (Mammalian Toxicology – Ecotoxicology joint session)19 in July 2019 and then at the TC 100 (Mammalian toxicology) and TC 104 (Ecotoxicology) in April 2023, and lastly in TC 163 in March 2025 after the submission of further data.20
With regard to the assessment of the ED potential of phenmedipham for humans and non‐target organisms according to the ECHA/EFSA guidance (2018), in determining whether phenmedipham interacts with the EAS‐ and thyroid (T)‐mediated pathways, the number and type of effects induced, and the magnitude and pattern of responses observed across studies were considered. Additionally, the conditions under which effects occur were considered, in particular, whether or not endocrine‐related responses occurred at dose(s) that also resulted in overt toxicity. The assessment is therefore providing a weight‐of‐evidence analysis of the potential interaction of phenmedipham with the EAS‐ and T‐signalling pathways using the available evidence in the data set.
T‐mediated parameters were considered sufficiently investigated and a consistent pattern of T‐mediated adversity was not identified in the available data set of mammalian studies.21 THs and thyroid‐stimulating hormone (TSH) analyses from the Extended‐One Generation Reproductive Toxicity (EOGRT) study were available and discussed at the Pesticide Peer Review meeting TC 163. Changes in THs and TSH levels were considered variable and not consistent throughout the different life stages tested in the EOGRT study (including, F0 and F1 parental, PND 4 and PND 21 offsprings). Phenmedipham tested positive in the TPO inhibition assay available in ToxCast (i.e. CCTE_Simmons_AUR_TPO_dn) with a potency comparable to the reference compound 6‐propyl‐2‐thiouracil (PTU); however, this finding was not supported by the in vivo evidence, i.e. lacking a clear pattern indicative of a sustained disruption of the hypothalamic–pituitary–thyroid (HPT) axis. Overall, based on the available and sufficient data set, a consistent pattern of T‐mediated adversity was not observed for phenmedipham. Therefore, it was concluded that the ED criteria are not met for the T‐modality (Scenario 1a of the ECHA/EFSA (2018) Guidance).
Concerning the EAS‐modalities, the data set was considered complete, and a pattern of EAS‐mediated adversity was not identified. Therefore, based on the available and sufficient data set, it was concluded that the ED criteria are not met for the EAS‐modalities (Scenario 1a of the EFSA/ECHA (2018) ED Guidance).
The outcome of the assessment reported above for humans also applies to wild mammals as non‐target organisms.
For non‐mammalian species, an extended amphibian metamorphosis assay22 (EAMA, in line with the protocol of Ortego et al., 2021) and a fish short‐term reproduction assay (FSTRA, OECD TG 229) were available and discussed at the Pesticides Peer‐Review meeting TC 104 and TC 163.23
In the available EAMA, at the highest tested concentration, a statistically significant delay in the time to reach metamorphosis, accompanied by histopathological changes in the thyroid (i.e. increase in follicular cell hyperplasia and hypertrophy) were observed in absence of systemic toxicity. In addition, positive evidence of T‐mediated endocrine activity (i.e. TPO inhibition) was observed in ToxCast (i.e. CCTE_Simmons_AUR_TPO_dn), supporting the findings observed in the available in‐vivo study. Therefore, based on the available data, i.e. positive evidence in vitro and in vivo and the biological plausible link between the endocrine activity (i.e. changes in thyroid histology) and the adversity (delay in time to reach NF 62), the overall Weight of Evidence pointed to a conclusion that phenmedipham is an ED through the T‐modality, although some uncertainties were identified (see table below).
Regarding the EAS‐modalities, decrease in fecundity accompanied by histopathological changes in the fish female gonads were seen in absence of systemic toxicity in the available FSTRA; however, no effects on VTG were observed and some of the changes observed in the gonad histopathology (i.e. decrease in post‐ovulatory follicles) are considered a direct consequence of the reduced fecundity. However, some sources of uncertainties were identified in the study and in the overall ED assessment (see Table 1 below). Overall, when considering the available findings and related uncertainties (see Table 1 below), no pattern of effects which can be related to a perturbation of the HPG axis was observed for the EAS‐modalities.
The uncertainties identified in the available studies and assessment, as summarised above, are not considered too high to prevent a conclusion on the endocrine disrupting properties of phenmedipham for non‐target organisms other than mammals through the EATS‐modalities.
Overall, it was concluded that phenmedipham does not meet the ED criteria for humans for EATS‐modalities as laid down in point 3.6.5. of Annex II to Regulation (EC) No 1107/2009, as amended by Commission Regulation (EU) 2018/605. For wild mammals as non‐target organisms, it can be concluded that phenmedipham does not meet the ED criteria for EATS‐modalities. For non‐target organisms other than mammals, phenmedipham does not meet the criteria through the EAS‐modalities; however, for the T‐modality for non‐target organisms other than mammals, it is concluded that phenmedipham meets the ED criteria as laid down in point 3.8.2 of Annex II to Regulation (EC) No 1107/2009, as amended by Commission Regulation (EU) 2018/605 (critical area of concern, see Section 10.2).
OVERVIEW OF THE RISK ASSESSMENT OF COMPOUNDS LISTED IN RESIDUE DEFINITIONS TRIGGERING ASSESSMENT OF EFFECTS DATA FOR THE ENVIRONMENTAL COMPARTMENTS (TABLES 2, 3, 4, 5)
7
PARTICULAR CONDITIONS PROPOSED TO BE TAKEN INTO ACCOUNT TO MANAGE THE RISK(S) IDENTIFIED
8
- No‐spray buffer zones and vegetated buffer strips up to 20 m are necessary to achieve low chronic risk to aquatic vertebrates (fish) in FOCUS Step 4 R3/stream scenario for phenmedipham.
- No‐spray buffer zones up to 10 m are necessary to achieve low chronic risk to aquatic invertebrates in FOCUS Step 4 D3/ditch and D4/stream scenarios for phenmedipham.
- No‐spray buffer zones and vegetated buffer strips up to 20 m are necessary to achieve low chronic risk to aquatic invertebrates in FOCUS Step 4 R1/stream for the use pattern of phenmedipham including two applications.
- No‐spray buffer zones and vegetated buffer strips up to 20 m are necessary to achieve low chronic risk to aquatic invertebrates in FOCUS Step 4 R/stream for the use pattern of phenmedipham including one application.
- No‐spray buffer zones and vegetated buffer strips up to 20 m are necessary to achieve a low acute risk to aquatic invertebrates in R1/stream and R3/stream scenarios for the metabolite 3‐methylaniline (m‐toluidine).
- No‐spray buffer zones and vegetated buffer strips up to 20 m are necessary to achieve a low chronic risk to aquatic invertebrates in R1/stream and R3/stream scenarios for the metabolite 3‐methylaniline (m‐toluidine).
DATA GAPS
9
This is a list of data gaps identified during the peer review process, including those areas in which a study may have been made available during the peer review process but not considered for procedural reasons (without prejudice to the provisions of Article 56 of Regulation (EC) No 1107/2009 concerning information on potentially harmful effects).
- A search of the scientific peer‐reviewed open literature on the active substance and its metabolites relevant to consumer exposure, dealing with side effects on health and published within the 10 years before the date of submission of the dossier, to be conducted and reported in accordance with EFSA guidance on the submission of scientific peer‐reviewed open literature for the approval of pesticide active substances under Regulation (EC) No 1107/2009 (EFSA, 2011). Details on the search performed on the active substance also need to be included in the RAR (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown, see Section 2).
- Spectra for identification of the relevant impurities (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 1).
- Content of relevant impurities, before and after storage (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 1).
- A method for determination of the relevant impurities in the representative formulation (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 1).
- Information on which analytical method has been used in each of the toxicological studies is missing (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 2).
- Toxicological information to address the toxicological relevance of most impurities present in the technical specifications from both applicants (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 2).
- Toxicokinetic data in rodents are needed to complete the toxicity profile of phenmedipham (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 2).
- Skin sensitisation study performed with the active substance (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 2).
- Data for the phototoxicity evaluation in the area of UVB wavelength (no validated method exists) (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 2).
- Acute toxicity studies on the representative plant protection product or equivalent formulation have not been provided (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 2).
- Genotoxicity data on plant/livestock metabolites 3‐aminophenol, 3‐acetamidophenol, 4‐acetamido‐o‐cresol, 4‐amino‐2‐methylphenol, acetamido‐benzoic acid, aminobenzoic acid and m‐acetotoluidine and, pending on further assessment in the residue section, repeated‐dose toxicity data relevant to consumer exposure may be needed. In addition, genotoxicity and repeated‐dose toxicity data relevant to consumer exposure are needed for 3‐methylaniline (m‐toluidine) (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Sections 2 and 3).
- Data to exclude the possible formation of MHPC and 3‐methylaniline (m‐toluidine) in sugar and sugar beets by‐products, used as a feed item for animal, has to be also investigated (relevant for the uses on sugar beet; submission date proposed by the applicant: unknown; see Section 3).
- Sufficient number of residue trials in sugar beet roots and leaves (4 NEU and 8 SEU) compliant with the representative GAPs and analysed according to the proposed residue definition for risk assessment have to be provided (relevant for all representative uses submission date proposed by the applicant: unknown; see Section 3).
- Animal feeding studies covered by valid storage stability data and analysed according to the residue definition for risk assessment for animal commodities, except poultry, (relevant for all representative uses; submission date proposed by the applicant: unknown; see Section 3).
- Information on the substances resulting from water treatment processes on the residues of phenmedipham (relevant for all representative uses evaluated; submission date proposed by the applicant: unknown; see Section 4).
- Further information to address the chronic risk to aquatic invertebrates from phenmedipham (relevant for the representative uses in sugar beet that include 2 or 3 applications at 320 g/ha; submission date proposed by the applicant: unknown; see Section 5).
- Further information to address the risk to the aquatic invertebrates from MHPC (relevant for all the representative uses; submission date proposed by the applicant: unknown; see Section 5).
- Further information to address the risk to the algae community from phenmedipham (relevant for all the representative uses; submission date proposed by the applicant: unknown; see Section 5).
- Further information to address the risk to bees from exposure via the puddle scenario (relevant for all representative uses; submission date proposed by the applicant: unknown; see Section 5).
- Further information to address the risk to bee larvae (relevant for all the representative uses in sugar beet; submission date proposed by the applicant: unknown; see Section 5).
- Further information to address the risk from sub‐lethal effects on bees (i.e. HPG) (relevant for all the representative uses in sugar beet; submission date proposed by the applicant: unknown; see Section 5).
CONCERNS
10
Issues that could not be finalised
10.1
An issue is listed as ‘could not be finalised’ if there is not enough information available to perform an assessment, even at the lowest tier level, for the representative uses in line with the uniform principles in accordance with Article 29(6) of Regulation (EC) No 1107/2009 and as set out in Commission Regulation (EU) No 546/201126 and if the issue is of such importance that it could, when finalised, become a concern (which would also be listed as a critical area of concern if it is of relevance to all representative uses).
An issue is also listed as ‘could not be finalised’ if the available information is considered insufficient to conclude on whether the active substance can be expected to meet the approval criteria provided for in Article 4 of Regulation (EC) No 1107/2009.
- The consumer risk assessment cannot be finalised due to incomplete data set from residue trials in sugar beet roots and leaves, the lack of investigation of the transfer of residues into animal commodities and missing toxicological data for several metabolites: 3‐aminophenol, 3‐acetamidophenol, 4‐acetamido‐o‐cresol, 4‐amino‐2‐methylphenol, aminobenzoic acid and m‐acetotoluidine (relevant for risk assessment residue definition for animal commodities) and 3‐methylaniline (relevant for processed commodities) (see Sections 2 and 3).
- Consumer risk assessment could not be finalised in relation to the substances resulting from water treatment processes on the residues of phenmedipham (see Sections 3 and 4).
- Aquatic risk assessment could not be finalised (algae) (see Section 5).
Critical areas of concern
10.2
An issue is listed as a critical area of concern if there is enough information available to perform an assessment for the representative uses in line with the uniform principles in accordance with Article 29(6) of Regulation (EC) No 1107/2009 and as set out in Commission Regulation (EU) No 546/2011, and if this assessment does not permit the conclusion that, for at least one of the representative uses, it may be expected that a plant protection product containing the active substance will not have any harmful effect on human or animal health or on groundwater, or any unacceptable influence on the environment.
An issue is also listed as a critical area of concern if the assessment at a higher tier level could not be finalised due to lack of information, and if the assessment performed at the lower tier level does not permit the conclusion that, for at least one of the representative uses, it may be expected that a plant protection product containing the active substance will not have any harmful effect on human or animal health or on groundwater, or any unacceptable influence on the environment.
An issue is also listed as a critical area of concern if, in the light of current scientific and technical knowledge using guidance documents available at the time of application, the active substance is not expected to meet the approval criteria provided for in Article 4 of Regulation (EC) No 1107/2009.
- 4The technical specifications proposed by either of the applicants, including the original one from Bayer are not covered by the batches used in the key toxicological studies27 (see Sections 2 and 5).
- 5For the T‐modality and non‐mammalian species, phenmedipham meets the criteria as laid down in point 3.8.2. of Annex II to Regulation (EC) No 1107/2009, as amended by Commission Regulation (EU) 2018/605 (see Section 6).
Overview of the concerns identified for each representative use considered
10.3
(If a particular condition proposed to be taken into account to manage an identified risk, as listed in Section 8, has been evaluated as being effective, then ‘risk identified’ is not indicated in Table 5.)
In addition to the issues identified in Sections 10.1 and 10.2, the technical material specification proposed was not comparable to the material used in the testing that would be used to derive the toxicological reference values. Also, for the T‐modality for non‐target organisms other than mammals, it is concluded that phenmedipham meets the ED criteria as laid down in point 3.8.2. of Annex II to Regulation (EC) No 1107/2009, as amended by Commission Regulation (EU) 2018/605 (Table 6).
ABBREVIATIONSAAOELacute acceptable operator exposure levelADIacceptable daily intakeAOELacceptable operator exposure levelAOPadverse outcome pathwayARapplied radioactivityARfDacute reference dosea.s.active substancebwbody weightCASChemical Abstracts ServiceDT_50_ period required for 50% dissipation (define method of estimation)EAMAextended amphibian metamorphosis assayECHAEuropean Chemicals AgencyEECEuropean Economic CommunityFAOFood and Agriculture Organization of the United NationsFIDflame ionisation detectorFIRfood intake rateFOBfunctional observation batteryFOCUSForum for the Co‐ordination of Pesticide Fate Models and their UseFSTRAfish short‐term reproduction assayGAPGood Agricultural PracticeGC–MSgas chromatography ‐ mass spectrometryHPGhypopharyngeal glandsHPThypothalamic–pituitary–thyroidIESTIinternational estimated short‐term intakeInChIKeyInternational Chemical Identifier KeyISOInternational Organization for StandardizationIUPACInternational Union of Pure and Applied ChemistryK_Foc_ Freundlich organic carbon adsorption coefficientLAGDAlarval amphibian growth and development assayLC–MS/MSliquid chromatography with tandem mass spectrometryLLNAlocal lymph node assayLOAELlowest observable adverse effect levelLOQlimit of quantificationMetHbmethaemoglobinMNmicronucleusMoAmechanism of actionMRLmaximum residue levelMSmass spectrometryMTCmaximum tolerated concentrationNEUnorthern EuropeNOAELno observed adverse effect levelOECDOrganisation for Economic Co‐operation and DevelopmentPBIplant‐back intervalPECpredicted environmental concentrationPOFpost ovulatory folliclesPTU6‐propyl‐2‐thiouracilQCquality controlQuEChERSQuick Easy Cheap Effective Rugged SafeRARrenewal assessment reportRP/HPLC‐UVreversed phase high performance liquid chromatography with UV detectorSCsuspension concentrateSEsuspoemulsionSEUsouthern EuropeSMILESsimplified molecular‐input line‐entry systemTHthyroid hormoneTPOthyroid peroxidaseTRRtotal radioactive residueTSHthyroid‐stimulating hormone (thyrotropin)UFuncertainty factorUVultravioletVTHvitellogeninWHOWorld Health OrganizationWoEweight of evidence
REQUESTOR
European Commission
QUESTION NUMBERS
EFSA‐Q‐2015‐00111, EFSA‐Q‐2019‐00025
COPYRIGHT FOR NON‐EFSA CONTENT
EFSA may include images or other content for which it does not hold copyright. In such cases, EFSA indicates the copyright holder and users should seek permission to reproduce the content from the original source.
NOTE/UPDATE
This scientific output, approved on 30 September 2025, supersedes the previous output published on 31 January 2018 (EFSA, 2018).
Supporting information
APPENDIX A: List of end points for phenmedipham and the formulation for representative uses
APPENDIX D: Non‐dietary exposure and consumer dietary exposure calculations
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1ECHA (European Chemicals Agency) . (2019). Committee for Risk Assessment (RAC) Opinion proposing harmonised classification and labelling at EU level of phenmedipham, EC Number: 237‐199‐0, CAS Number: 13684–63‐4. Adopted 20 September 2019. CLH‐O‐0000001412‐86‐297/F. https://www.echa.europa.eu/documents/10162/5cf 0db 4f‐b 6c 0‐ebd 0‐a 40d‐8b 9859 b 28c 96
- 2ECHA and EFSA (European Chemicals Agency and European Food Safety Authority) with the technical support of the Joint Research Centre (JRC) , Andersson, N. , Arena, M. , Auteri, D. , Barmaz, S. , Grignard, E. , Kienzler, A. , Lepper, P. , Lostia, A. M. , Munn, S. , Parra Morte, J. M. , Pellizzato, F. , Tarazona, J. , Terron, A. , & Van der Linden, S. (2018). Guidance for the identification of endocrine disruptors in the context of regulations (EU) No 528/2012 and (EC) No 1107/2009. · doi ↗ · pubmed ↗
- 3EFSA (European Food Safety Authority) . (2009). Guidance on risk assessment for birds and mammals on request from EFSA. EFSA Journal, 7(12), 1438. 10.2903/j.efsa.2009.1438 40123698 PMC 11926626 · doi ↗ · pubmed ↗
- 4EFSA (European Food Safety Authority) . (2011). Submission of scientific peer‐reviewed open literature for the approval of pesticide active substances under regulation (EC) No 1107/2009. EFSA Journal, 9(2), 2092. 10.2903/j.efsa.2011.2092 · doi ↗
- 5EFSA (European Food Safety Authority) . (2013). EFSA Guidance document on the risk assessment of plant protection products on bees (Apis mellifera, Bombus spp. and solitary bees). EFSA Journal, 11(7), 3295. 10.2903/j.efsa.2013.3295 PMC 1017385237179655 · doi ↗ · pubmed ↗
- 6EFSA (European Food Safety Authority) . (2014 a). Reasoned opinion on the review of the existing maximum residue levels (MR Ls) for phenmedipham according to article 12 of regulation (EC) No 396/2005. EFSA Journal, 12(8), 3807. 10.2903/j.efsa.2014.3807 PMC 1188313440061611 · doi ↗ · pubmed ↗
- 7EFSA (European Food Safety Authority) . (2014 b). Guidance on the assessment of exposure of operators, workers, residents and bystanders in risk assessment for plant protection products. EFSA Journal, 12(10), 3874. 10.2903/j.efsa.2014.3874 PMC 876509135079284 · doi ↗ · pubmed ↗
- 8EFSA (European Food Safety Authority) . (2017). Peer review report to the conclusion regarding the peer review of the pesticide risk assessment of the active substance phenmedipham. www.efsa.europa.eu 10.2903/j.efsa.2018.5151 PMC 700951032625695 · doi ↗ · pubmed ↗