p62-dependent caspase-2 activation governs TDP-43 clearance and neuronal fate in ALS
Pavel I. Volik, Gelina S. Kopeina, Boris Zhivotovsky, Alexey V. Zamaraev

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
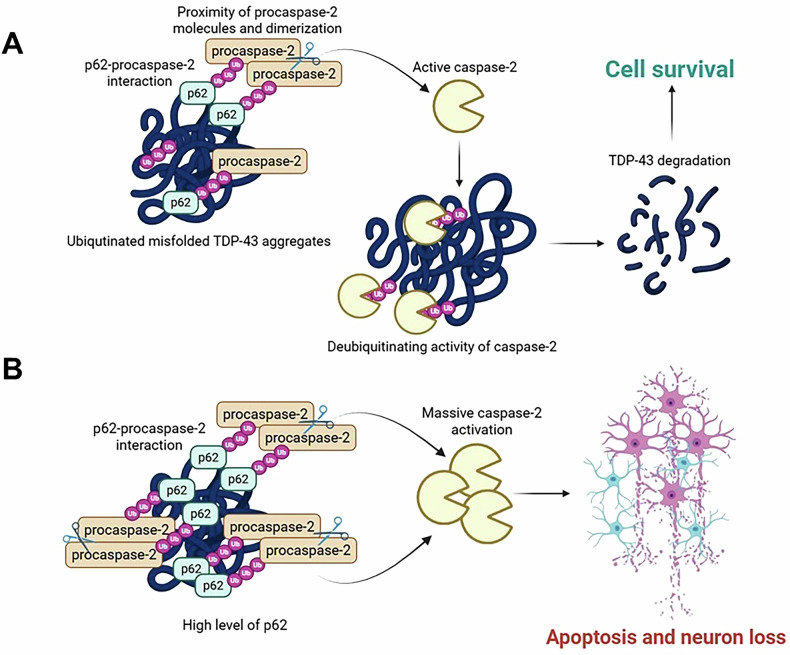 Figure 1
Figure 1- —https://doi.org/10.13039/501100002794Cancerfonden (Swedish Cancer Society)
- —https://doi.org/10.13039/501100007232Radiumhemmets Forskningsfonder (Cancer Research Foundations of Radiumhemmet)
- —https://doi.org/10.13039/501100006769Russian Science Foundation (RSF)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyotrophic Lateral Sclerosis Research · Genetic Neurodegenerative Diseases · Tuberous Sclerosis Complex Research
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by a loss of motor neurons. Progressive muscle weakness leads to the incremental development of respiratory failure and subsequent death of ALS patients 2–3 years after symptom onset. A central pathological hallmark of ALS is the formation of misfolded protein aggregates in degenerating motor neurons and surrounding oligodendrocytes [1]. However, the mechanism by which these cytoplasmic inclusions or aggregates affect neuronal function and cause motor neuron death remains poorly understood.
The majority of ALS cases are sporadic (sALS), while 5–10% are familial in origin (fALS). In 1993, Cu/Zn-binding superoxide dismutase (SOD1) was identified as the first protein aggregated in fALS due to the SOD1 gene missense mutation. Since then, several novel ALS-associated gene defects have been identified, including mutations in TARDBP, the gene encoding TAR DNA-binding protein 43 kDa (TDP-43), a highly conserved DNA/RNA-binding protein that preferentially recognizes UG-rich and TG-rich motifs of RNA and DNA. TDP-43 has been reported as a prominent component of ubiquitinated and aberrantly phosphorylated cytosolic protein aggregates detected in 97% of all ALS patients, regardless of the mechanisms of disease onset. More than 50 mutations in TARDBP have been associated with ALS, confirming a crucial role of TDP-43 in ALS pathology [2]. N-terminally truncated forms of TDP-43, C-terminal fragments 35 kDa (CTF35) and 25 kDa (CTF25), are typically observed in ALS aggregations and associated with cell toxicity leading to the generation of insoluble ubiquitin- and phospho-positive cytoplasmic inclusions. TDP-43 CTFs could be generated by translation of alternatively spliced isoforms or proteolytic cleavage of full-length TDP-43. CTF35 and CTF25 are generated through proteolytic cleavage induced by caspase-3, -4 and -7, while calpains and δ-secretase can also cleave TDP-43, leading to CTFs’ formation. Other lower-abundance truncated fragments of TDP-43 (15–16 kDa, 22–25 kDa, and 33–37 kDa) have also been identified in ALS, but their role in disease pathology remains unclear [2].
Due to the cytotoxicity of TDP-43 species, the clearance of TDP-43 is a crucial cell survival mechanism. The degradation of full-length and CTFs of TDP-43 is normally mediated by the ubiquitin-proteasome system, which is disrupted by ALS-related mutations in the gene encoding Ubiquilin-2. Suppression of this clearance pathway stimulates a massive accumulation of TDP-43 aggregates in the cytoplasm of primary neurons [1]. Autophagy represents another critical clearance mechanism. The gain-of-function mutation (c.2155 A > G,p.M719V) in the deubiquitinase CYLD reduces autophagy, which correlates with TDP-43 mislocalization and aggregation [2]. Furthermore, TDP-43 aggregates, comprising full-length and CTFs of TDP-43, colocalize with autophagy-related protein sequestosome 1 (SQSTM1/p62). Previously, p62 was reported to decrease TDP-43 aggregation by promoting its autophagy- and proteasome-dependent degradation [3]. SQSTM1 overexpression has recently been found to promote CTF25 clearance in monkey substantia nigra [4]. Moreover, the identification of several pathogenic SQSTM1 mutations in patients with ALS supports the involvement of p62 in TDP-43 clearance [5]. MicroRNA-183-5p, which suppresses SQSTM1 expression, resulting in TARDBP upregulation and increased TDP-43 aggregation, can regulate the p62-dependent degradation of TDP-43 [6]. However, recent findings have indicated that p62 may play a role in the accumulation of toxic, misfolded fragments of TDP-43 in the cytoplasm. Nevertheless, the duration of experiments monitoring TDP-43 turnover in that study may provide a rationale for this controversial effect [7].
In ALS, aggregates of TDP-43 are characterized by excessive accumulation of post-translational modifications, specifically phosphorylation, ubiquitination, and poly-ubiquitination [2]. A recent study [8] revealed that caspase-2, a multifunctional cysteine protease involved in cell death, genomic stability, and the oxidative stress response, could bind to poly-ubiquitinated conjugates via its allosteric ubiquitin-interacting motif-like region. It removes overloaded ubiquitin chains in a protease-dependent manner to mediate condensate disassembly and the degradation of misfolded proteins. The deficiency of CASP2 results in the accumulation of stress-induced ubiquitinated complexes and pathological poly-ubiquitinated TDP-43 aggregates, promoting neuromuscular denervation in mice [9]. However, it is unclear how caspase-2 is activated in this scenario, since the enzyme is normally present in cells in the form of an inactive zymogen known as procaspase-2. The mechanism of caspase-2 activation is closely related to the induction of spatial proximity of procaspase-2 molecules, their dimerization, and subsequent autoproteolysis with active caspase-2 release. Recent findings unexpectedly demonstrate that caspase-2 can be similarly activated via ubiquitin-dependent interaction with p62 [10]. The abundance of p62 observed in ubiquitinated TDP-43-positive aggregates could promote proximity, dimerization, and autocleavage of procaspase-2 molecules, leading to the formation of active caspase-2, which can promote TDP-43 degradation, acting as a deubiquitinase. Thus, p62 could contribute to TDP-43 aggregates degradation through caspase-2 activation, preventing the development of ALS pathology (Fig. 1A).Fig. 1. The mode of TDP-43 clearance by p62.A Activation of caspase-2 facilitates the removal of overloaded ubiquitin chains on TDP-43 prone to misfolding. B Caspase-2 switches to an apoptotic function in the presence of high p62 levels and excessive accumulation of TDP-43 inclusions.
Nevertheless, an important question related to the apoptotic function of caspase-2, activated by p62, remains. Under physiological conditions, the level of active caspase-2 molecules is sufficient to eliminate misfolded poly-ubiquitinated aggregates via deubiquitinase activity, but it is insufficient to induce neuronal apoptosis. However, in ALS pathology, excessive TDP-43 accumulation overwhelms these degradation pathways. The excessive accumulation of insoluble ubiquitinated p62-containing TDP-43 aggregates, whose clearance is counteracted, could promote intense caspase-2 activation through proximity-induced dimerization, switching its role from that of a proteostasis regulator to that of an apoptotic initiator, thereby eliminating neurons with irreversible proteostatic collapse (Fig. 1B). The fact that SQSTM1 overexpression accelerates ALS onset and shortens the lifespan of a mutant SOD1^H46R^-tg ALS mouse model supports this hypothesis [11]. Furthermore, a negative association of p62 levels with motor neuron loss in the spinal cord and disease duration was also found in patients with sporadic ALS [12]. These findings suggest dual context-dependent functions for caspase-2 in ALS and support further investigation of therapeutic strategies that modulate its activity to delay ALS progression.