The chromosomal genome sequence of the sponge, Corticium candelabrum Schmidt, 1862 and its associated microbial metagenome sequences
Manuel Maldonado, Lucia Pita, Dirk Erpenbeck, Ute Hentschel, Graeme Oatley, Elizabeth Sinclair, Eerik Aunin, Noah Gettle, Camilla Santos, Michael Paulini, Haoyu Niu, Victoria McKenna, Rebecca O’Brien, Richard R Copley, Jayan Duminda M Senevirathna, Estelle Proux-Wéra

TL;DR
This paper presents the genome and microbial metagenome of the sponge Corticium candelabrum, including detailed gene annotations and bacterial genomes.
Contribution
The study provides a high-quality chromosomal genome assembly and metagenome of a sponge species, including 44 high-quality bacterial MAGs.
Findings
The sponge genome is 185.49 megabases long with 26,198 protein-coding genes.
The mitochondrial genome is 18.19 kilobases and 99.4% of the assembly is scaffolded into 22 chromosomal pseudomolecules.
53 bacterial genomes were identified, including 44 high-quality MAGs from diverse phyla.
Abstract
We present a genome assembly from a specimen of Corticium candelabrum (sponge; Porifera; Homoscleromorpha; Homosclerophorida; Plakinidae). The genome sequence has a total length of 185.49 megabases. Most of the assembly (99.4%) is scaffolded into 22 chromosomal pseudomolecules. The mitochondrial genome has also been assembled and is 18.19 kilobases in length. Gene annotation of this assembly on Ensembl identified 26,198 protein-coding genes. The metagenome of the specimen was also assembled, and 53 binned bacterial genomes were identified, including 44 high-quality MAGs that were typical of high microbial abundance sponge and included, besides the phyla Chloroflexota (class Dehalococcoidia), Acidobacteriota (order Acidomicrobiales), Alpha- and Gammaproteobacteria, also representatives of several candidatus phyla (Candidatus Latescibacterota, Binatota, Poribacteria)
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1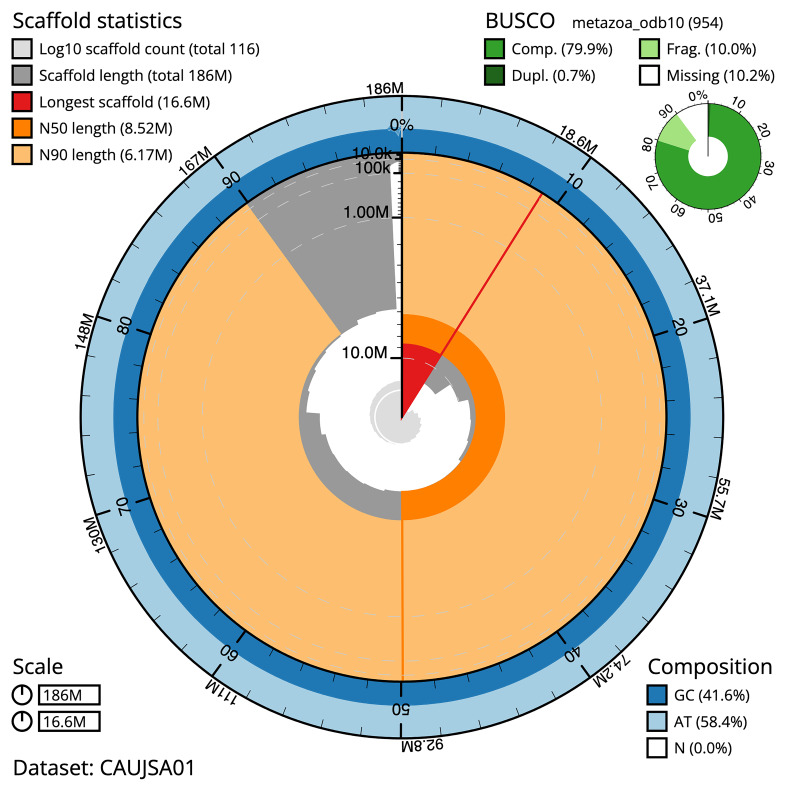 Figure 2
Figure 2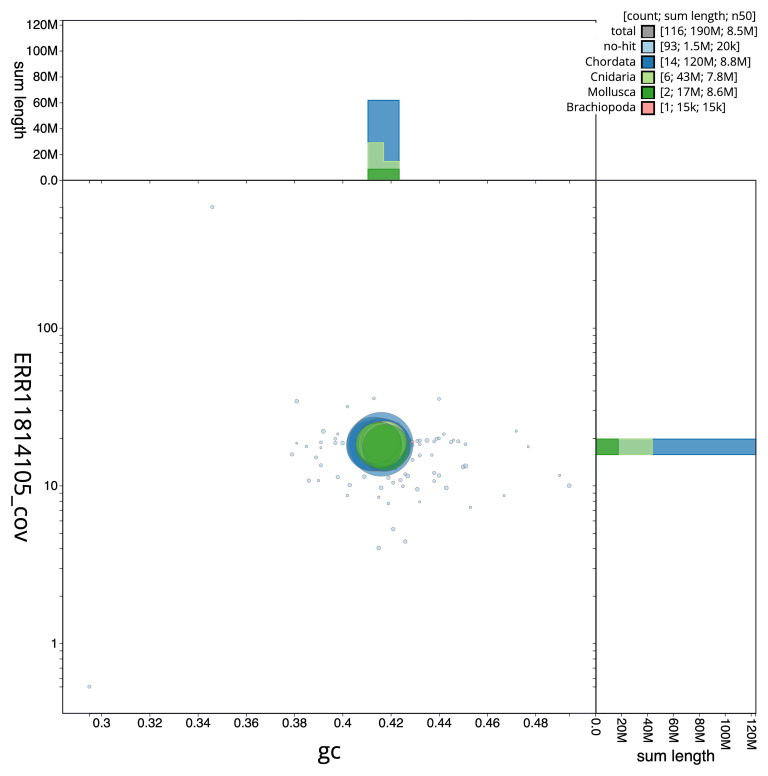 Figure 3
Figure 3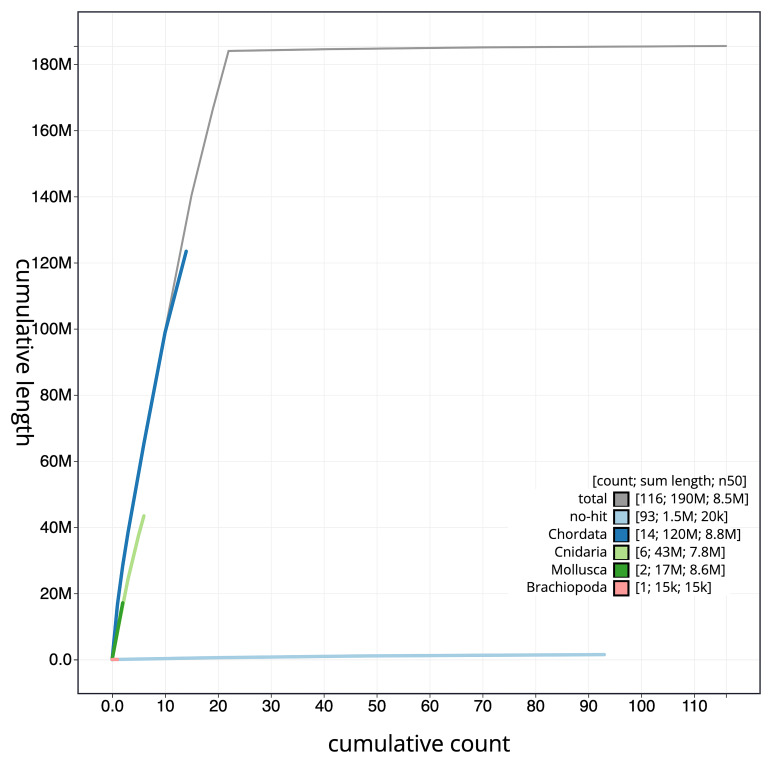 Figure 4
Figure 4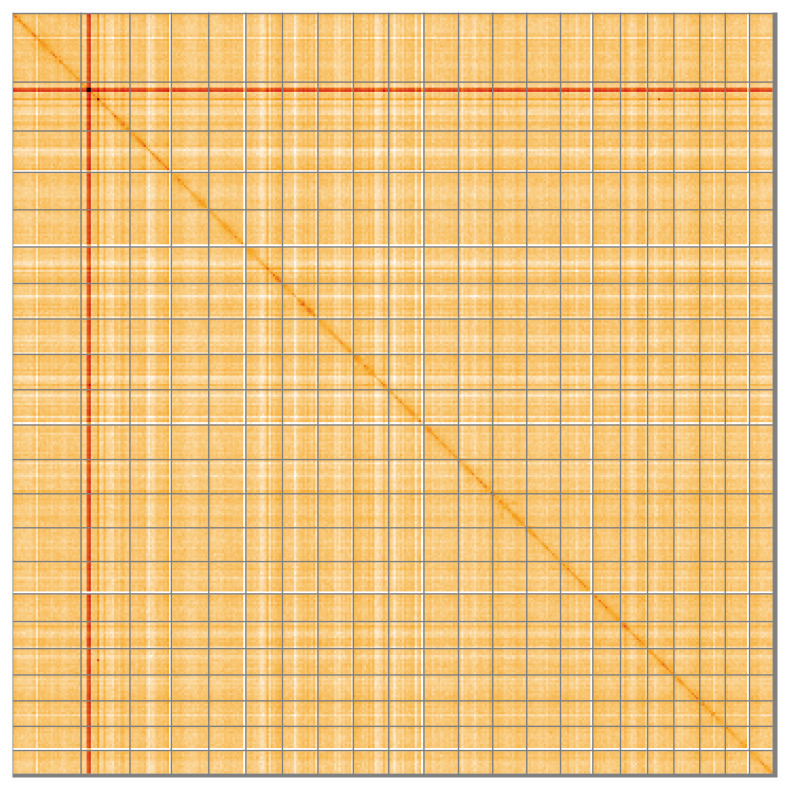 Figure 5
Figure 5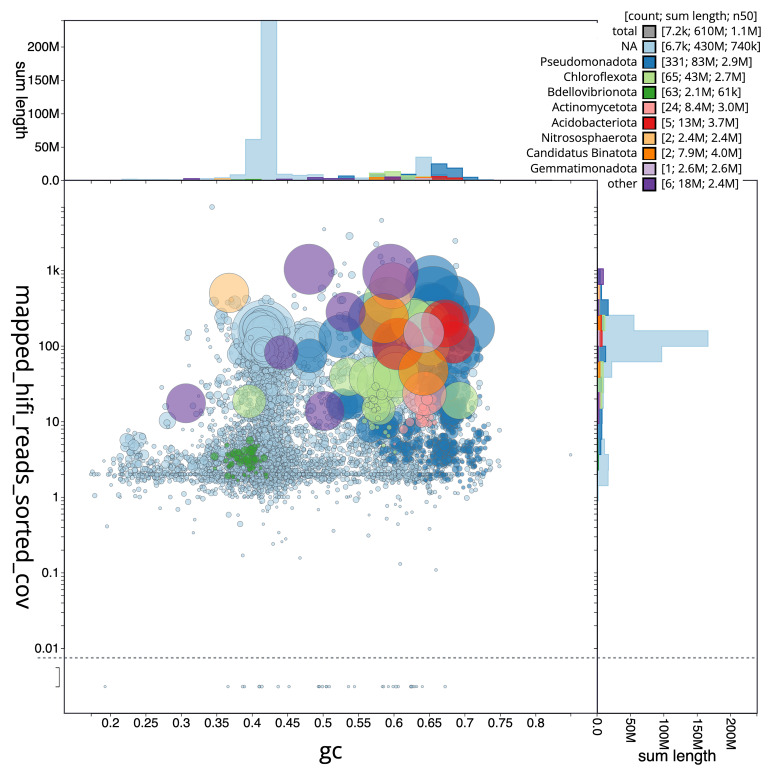 Figure 6
Figure 6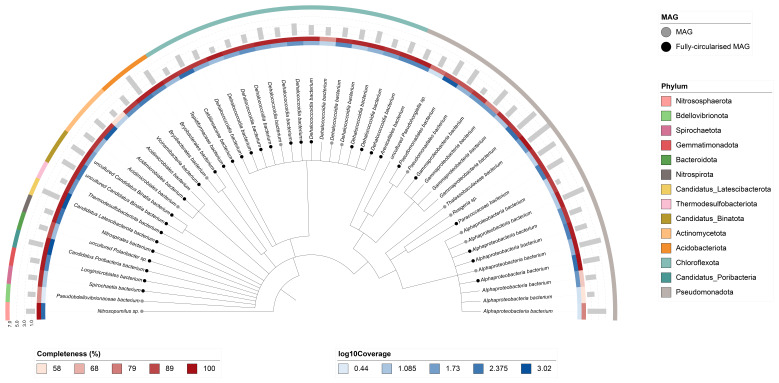 Figure 7
Figure 7| Project information | |||
|---|---|---|---|
|
| Corticium candelabrum | ||
|
| PRJEB64714 | ||
|
|
| ||
|
| SAMEA9361900 | ||
|
| 121492 | ||
| Specimen information | |||
|
|
|
|
|
|
| ooCorCand1 | SAMEA9361972 | Somatic tissue |
|
| ooCorCand1 | SAMEA9361954 | Somatic tissue |
|
| ooCorCand1 | SAMEA9361956 | Somatic tissue |
| Sequencing information | |||
|
|
|
|
|
|
| ERR11814104 | 9.45e+08 | 142.68 |
|
| ERR11809138 | 2.40e+06 | 28.7 |
|
| ERR11809139 | 1.81e+06 | 17.65 |
|
| ERR14749924 | 1.98e+06 | 20.57 |
|
| ERR11814103 | 3.65e+07 | 5.52 |
|
| ERR14986693 | 8.13e+07 | 12.28 |
| Genome assembly | |
|---|---|
| Assembly name | ooCorCand1.1 |
| Assembly accession | GCA_963422355.1 |
|
|
|
| Assembly level for primary assembly | chromosome |
| Span (Mb) | 185.49 |
| Number of contigs | 392 |
| Number of scaffolds | 115 |
| Longest scaffold (Mb) | 16.58 |
| Assembly metric | Measure |
| Contig N50 length | 1.3 Mb |
| Scaffold N50 length | 8.52 Mb |
| Consensus quality (QV) | Primary: 52.3; alternate: 52.4; combined 52.3 |
| BUSCO
| C:79.9%[S:79.1%,D:0.7%],F:10.0%,M:10.2%,n:954 |
| Percentage of assembly assigned to chromosomes | 99.2% |
| Organelles | Mitochondrial genome: 18.19 kb |
| Genome annotation of assembly GCA_963422355.1 at Ensembl | |
| Number of protein-coding genes | 26,198 |
| Number of non-coding genes | 413 |
| Number of gene transcripts | 40,526 |
| INSDC accession | Name | Length (Mb) | GC% |
|---|---|---|---|
| 1 | 16.58 | 41.5 | |
| 2 | 11.83 | 41.5 | |
| 3 | 10.01 | 41 | |
| 4 | 9.05 | 42 | |
| 5 | 8.98 | 41.5 | |
| 6 | 8.85 | 41.5 | |
| 7 | 8.55 | 41.5 | |
| 8 | 8.58 | 42 | |
| 9 | 8.57 | 41.5 | |
| 10 | 8.52 | 41.5 | |
| 11 | 8.36 | 41.5 | |
| 12 | 8.22 | 42 | |
| 13 | 8.28 | 41.5 | |
| 14 | 8.17 | 41.5 | |
| 15 | 7.82 | 41.5 | |
| 16 | 6.7 | 41.5 | |
| 17 | 6.54 | 41.5 | |
| 18 | 6.37 | 42 | |
| 19 | 6.26 | 42 | |
| 20 | 6.17 | 42 | |
| 21 | 5.85 | 41.5 | |
| 22 | 5.74 | 41.5 | |
| MT | 0.02 | 34.5 |
| Software tool | Version | Source |
|---|---|---|
| BEDTools | 2.30.0 |
|
| bin3C | 0.3.3 |
|
| BLAST | 2.14.0 |
|
| BlobToolKit | 4.2.1 |
|
| BUSCO | 5.3.2 |
|
| bwa-mem2 | 2.2.1 |
|
| CheckM | 1.2.1 |
|
| Cooler | 0.8.11 |
|
| DIAMOND | 2.0.15 |
|
| dRep | 3.4.0 |
|
| fasta_windows | 0.2.4 |
|
| FastK | 1.1 |
|
| gEVAL | N/A |
|
| Gfastats | 1.3.6 |
|
| GoaT CLI | 0.2.5 |
|
| GTDB-TK | 2.3.2 |
|
| Hifiasm | 0.16.1 |
|
| HiGlass | 1.13.4 |
|
| MAGScoT | 1.0.0 |
|
| MaxBin | 2.7 |
|
| MerquryFK | 1.1 |
|
| MetaBat2 | 2.15-15-gd6ea400 |
|
| metaMDBG | Pre-release |
|
| MetaTOR | - |
|
| Minimap2 | 2.24-r1122 |
|
| MitoHiFi | 2 |
|
| MultiQC | 1.14, 1.17, and 1.18 |
|
| PretextView | 0.2.5 |
|
| PROKKA | 1.14.5 |
|
| purge_dups | 1.2.3 |
|
| samtools | 1.15.1 |
|
| Seqtk | 1.3 |
|
| Singularity | 3.9.0 |
|
| YaHS | 1.1a.2 |
|
- —Wellcome Trust
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMarine Sponges and Natural Products · Genomics and Phylogenetic Studies · Microbial Natural Products and Biosynthesis
Species taxonomy
Eukaryota; Opisthokonta; Metazoa; Porifera; Homoscleromorpha; Homosclerophorida; Plakinidae; Corticium; Corticium candelabrum Schmidt, 1862 (NCBI:txid121492)
Background
Corticium candelabrum Schmidt, 1862 is a sponge species originally described from the sublittoral rocky bottoms of the Mediterranean region, where it is common. The species has also widely been reported from several Caribbean locations, the Atlantic Coast of Canada and Spain, the Indian Ocean, Indonesia, Papua New Guinea, New Zealand, and around Australia ( de Voogd et al., 2024). The material used for this genome sequencing and the current species description are both derived from populations in the Western Mediterranean, the biogeographic region where the sponge was first described.
Corticium candelabrum is a light brown sponge that grows as lobate, cerebriform, plate-like individuals, reaching up to 10–12 cm in largest diameter and no more than 0.5 cm in thickness ( Figure 1). It has a smooth surface, with small osculi and ostia visible to the naked eye. The sponge typically inhabits rocky walls and overhangs and has no known predators other than the nudibranch Platydoris argo ( De Caralt et al., 2007).
Corticium candelabrum specimen used for genome sequencing, photographed in the CEAB wet laboratory three hours after collection and immediately prior to tissue dissection.Underwater photography by Manuel Maldonado.
The species has notably helped to better understand the evolution of Porifera in several respects. This species was shown to have a basement membrane of type IV collagen beneath some epithelia, a condition typical of bilaterian metazoans and found only in sponges of the classes Homoscleromorpha and Calcarea ( Boute et al., 1996; Wörheide et al., 2012). The analysis of previous transcriptomes and the current genome has allowed to establish that Class Homoscleromorpha has independently evolved its own protein machineries for both transporting silicon and for making siliceous spicules, which are phylogenetically unrelated to those in the classes Demospongiae and Hexactinellida (Leria & Maldonado, under review; Maldonado et al., under review; Shimizu et al., 2024).
Corticium candelabrum is a hermaphroditic species that undergoes internal fertilisation and broods its embryos until the release of a cinctogastrula larvae ( Maldonado & Abdul Wahab, 2025; Maldonado & Riesgo, 2008). In Western Mediterranean populations, larval release occurs from mid-June to late-July, with dense populations producing an estimated half a million larvae per square metre annually ( Maldonado & Riesgo, 2008). The whitish larva is about 350 µm in length. It is entirely ciliated and swims for several days in the water column before settling and metamorphosing into a small juvenile sponge.
The microbiome of Corticium candelabrum is characteristic of high microbial abundance sponges, including ammonia-oxidising archaea and bacteria such as Alpha- and Gammaproteobacteria, Actinobacteria, Candidatus phylum Poribacteria, Nitrospira, and Chloroflexi ( Sharp et al., 2007; Sipkema et al., 2015). The process of vertical microbial transmission is well documented through electron microscopy studies ( De Caralt et al., 2007; Maldonado, 2007; Riesgo et al., 2007). Dense and diverse extracellular microbes are located particularly in subepithelial regions of the mesohyl and around oocytes and embryos ( Maldonado, 2007). The microbes surrounding the embryos migrate on their own and infiltrate the intercellular spaces that are formed between the blastomeres during the process of embryonic segmentation. As embryonic development progresses, a major cellular reorganisation occurs in a process analogous to gastrulation, wherein internal blastomeres migrate toward the periphery, leaving a central cavity, where all the microbes that had entered the embryo become finally enclosed.
The whole-genome sequencing of C. candelabrum provides a valuable tool to investigate the genomic underpinnings of its biology, ecology, and evolution. This genome is especially significant for understanding the evolutionary emergence of sponge classes, particularly the divergence between the Hexactinellida-Demospongiae and Homoscleromorpha-Calcarea clades. It also provides insights into the evolution of the siliceous skeleton of Porifera, as well as the origins of silicon biomineralization in Metazoa.
Genome sequence report
Sequencing data
The genome of a specimen of Corticium candelabrum ( Figure 1) was sequenced using Pacific Biosciences single-molecule HiFi long reads, generating 66.92 Gb from 6.19 million reads. Based on the estimated genome size, the sequencing data provided approximately 70 coverage of the genome. Chromosome conformation Hi-C data produced 142.68 Gb from 944.91 million reads. RNA sequencing data were also generated and are available in public sequence repositories. Table 1 summarises the specimen and sequencing information.
Table 1.: Specimen and sequencing data for Corticium candelabrum.
Assembly statistics
The primary haplotype was assembled, and contigs corresponding to an alternate haplotype were also deposited in INSDC databases. The assembly was improved by manual curation, which corrected 55 misjoins or missing joins and removed one haplotypic duplication. These interventions decreased the scaffold count by 9.38% and the scaffold N50 by 40.2%. The final assembly has a total length of 185.49 Mb in 115 scaffolds, with 277 gaps, and a scaffold N50 of 8.52 Mb ( Table 2).
Table 2.: Genome assembly data for Corticium candelabrum.
The snail plot in Figure 2 provides a summary of the assembly statistics, indicating the distribution of scaffold lengths and other assembly metrics. Figure 3 shows the distribution of scaffolds by GC proportion and coverage. Figure 4 presents a cumulative assembly plot, with separate curves representing different scaffold subsets assigned to various phyla, illustrating the completeness of the assembly.
Genome assembly of Corticium candelabrum, ooCorCand1.1: metrics.The BlobToolKit snail plot provides an overview of assembly metrics and BUSCO gene completeness. The circumference represents the length of the whole genome sequence, and the main plot is divided into 1,000 bins around the circumference. The outermost blue tracks display the distribution of GC, AT, and N percentages across the bins. Scaffolds are arranged clockwise from longest to shortest and are depicted in dark grey. The longest scaffold is indicated by the red arc, and the deeper orange and pale orange arcs represent the N50 and N90 lengths. A light grey spiral at the centre shows the cumulative scaffold count on a logarithmic scale. A summary of complete, fragmented, duplicated, and missing BUSCO genes in the metazoa_odb10 set is presented at the top right. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/Corticium%20candelabrum/dataset/CAUJSA01/snail.
Genome assembly of Corticium candelabrum, ooCorCand1.1: BlobToolKit GC-coverage plot.Blob plot showing sequence coverage (vertical axis) and GC content (horizontal axis). The circles represent scaffolds, with the size proportional to scaffold length and the colour representing phylum membership. The histograms along the axes display the total length of sequences distributed across different levels of coverage and GC content. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/Corticium%20candelabrum/dataset/CAUJSA01/blob.
Genome assembly of Corticium candelabrum, ooCorCand1.1: BlobToolKit cumulative sequence plot.The grey line shows cumulative length for all scaffolds. Coloured lines show cumulative lengths of scaffolds assigned to each phylum using the buscogenes taxrule. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/Corticium%20candelabrum/dataset/CAUJSA01/cumulative.
Most of the assembly sequence (99.2%) was assigned to 22 chromosomal-level scaffolds. These chromosome-level scaffolds, confirmed by Hi-C data, are named according to size ( Figure 5; Table 3).
Genome assembly of Corticium candelabrum: Hi-C contact map of the ooCorCand1.1 assembly, visualised using HiGlass.Chromosomes are shown in order of size from left to right and top to bottom. An interactive version of this figure is available at https://genome-note-higlass.tol.sanger.ac.uk/l/?d=WjDYLGxASlOVoIXW1QIYRw.
Table 3.: Chromosomal pseudomolecules in the genome assembly of Corticium candelabrum, ooCorCand1.
The mitochondrial genome was also assembled. This sequence is included as a contig in the multifasta file of the genome submission and as a standalone record in GenBank.
Assembly quality metrics
The combined primary and alternate assemblies achieve an estimated QV of 52.3. BUSCO v. 5.3.2 analysis using the metazoa_odb10 reference set ( n = 954) identified 79.9% of the expected gene set (single = 79.1%, duplicated = 0.7%).
Metagenome report
Fifty-three binned genomes were generated from the metagenome assembly ( Figure 6), of which 44 were classified as high-quality metagenome assembled genomes (MAGs) (see methods). The completeness values for these assemblies range from approximately 58% to 100% with contamination below 9%. A cladogram of the binned metagenomes is shown in Figure 7. (For details on binned genomes see https://doi.org/10.6084/m9.figshare.30017644.)
Blob plot of base coverage mapped against GC proportion for sequences in the metagenome of Corticium candelabrum.Binned metagenomes are coloured by phylum. Circles are sized in proportion to sequence length on a square root scale, ranging from 501 to 6,575,414. Histograms show the distribution of sequence length sum along each axis. An interactive version of this figure may be viewed here.
Cladogram showing the taxonomic placement of metagenome bins, constructed using NCBI taxonomic identifiers with taxonomizr and annotated in iTOL.Colours indicate phylum-level taxonomy. Additional tracks show sequencing coverage (log₁₀), genome size (Mbp), and completeness. Bins that meet the criteria for MAGs are marked with a grey circle; fully circularised MAGs are marked in black.
Genome annotation report
The Corticium candelabrum genome assembly (GCA_963422355.1) was annotated at the European Bioinformatics Institute (EBI) using the Ensembl Genebuild pipeline. The resulting annotation includes 40,526 transcribed mRNAs from 26,198 protein-coding and 413 non-coding genes ( Table 2; https://beta.ensembl.org/species/9d1ccad7-a202-4858-9275-8224ad9515df). The average transcript length is 5,686.84, with an average of 1.52 coding transcripts per gene and 6.81 exons per transcript.
Methods
Sample acquisition
An adult Corticium candelabrum (specimen ID GHC0000185, ToLID ooCorCand1) was collected from Blanes, Girona, Spain (latitude 41.67333, longitude 2.80314) on 2021-02-01 by scuba diving. The specimen was collected and identified by Manuel Maldonado (CEAB-CSIC) and preserved by snap-freezing.
Nucleic acid extraction
The workflow for high molecular weight (HMW) DNA extraction at the Wellcome Sanger Institute (WSI) Tree of Life Core Laboratory includes a sequence of procedures: sample preparation and homogenisation, DNA extraction, fragmentation and purification. Detailed protocols are available on protocols.io ( Denton et al., 2023). The ooCorCand1 sample was prepared for DNA extraction by weighing and dissecting it on dry ice ( Jay et al., 2023). Tissue was cryogenically disrupted using the Covaris cryoPREP ^®^ Automated Dry Pulverizer ( Narváez-Gómez et al., 2023). HMW DNA was extracted using the Manual MagAttract v1 protocol ( Strickland et al., 2023b). DNA was sheared into an average fragment size of 12–20 kb in a Megaruptor 3 system ( Todorovic et al., 2023). Sheared DNA was purified by solid-phase reversible immobilisation, using AMPure PB beads to eliminate shorter fragments and concentrate the DNA ( Strickland et al., 2023a). The concentration of the sheared and purified DNA was assessed using a Nanodrop spectrophotometer and Qubit Fluorometer using the Qubit dsDNA High Sensitivity Assay kit. Fragment size distribution was evaluated by running the sample on the FemtoPulse system.
RNA was also extracted from tissue of ooCorCand1 in the Tree of Life Laboratory at the WSI using the RNA Extraction: Automated MagMax™ mirVana protocol ( do Amaral et al., 2023). The RNA concentration was assessed using a Nanodrop spectrophotometer and a Qubit Fluorometer using the Qubit RNA Broad-Range Assay kit. Analysis of the integrity of the RNA was done using the Agilent RNA 6000 Pico Kit and Eukaryotic Total RNA assay.
Sequencing
Pacific Biosciences HiFi circular consensus DNA sequencing libraries were constructed according to the manufacturers’ instructions. DNA sequencing was performed by the Scientific Operations core at the WSI on the Pacific Biosciences Sequel IIe instrument. Tissue from the ooCorCand1 sample was processed for Hi-C sequencing at the WSI Scientific Operations core, using the Arima-HiC v2 kit and sequenced on the Illumina NovaSeq 6000 instrument. Poly(A) RNA-Seq libraries were constructed using the NEB Ultra II RNA Library Prep kit, following the manufacturer’s instructions. RNA sequencing was performed on the Illumina NovaSeq X instrument.
Genome assembly, curation and evaluation
** Assembly **
Prior to assembly of the PacBio HiFi reads, a database of k-mer counts ( k = 31) was generated from the filtered reads using FastK. GenomeScope2 ( Ranallo-Benavidez et al., 2020) was used to analyse the k-mer frequency distributions, providing estimates of genome size, heterozygosity, and repeat content.
The HiFi reads were assembled using Hifiasm ( Cheng et al., 2021) with the --primary option. Haplotypic duplications were identified and removed using purge_dups ( Guan et al., 2020). The Hi-C reads were mapped to the primary contigs using bwa-mem2 ( Vasimuddin et al., 2019). The contigs were further scaffolded using the provided Hi-C data ( Rao et al., 2014) in YaHS ( Zhou et al., 2023) using the --break option for handling potential misassemblies. The scaffolded assemblies were evaluated using Gfastats ( Formenti et al., 2022), BUSCO ( Manni et al., 2021) and MERQURY.FK ( Rhie et al., 2020).
The mitochondrial genome was assembled using MitoHiFi ( Uliano-Silva et al., 2023), which runs MitoFinder ( Allio et al., 2020) and uses these annotations to select the final mitochondrial contig and to ensure the general quality of the sequence.
** Assembly curation **
The assembly was checked for contamination and corrected using the gEVAL system ( Chow et al., 2016) as described previously ( Howe et al., 2021). Manual curation was conducted primarily in PretextView ( Harry, 2022) and HiGlass ( Kerpedjiev et al., 2018). Any identified contamination, missed joins, and mis-joins were amended, and duplicate sequences were tagged and removed. The curation process is documented at https://gitlab.com/wtsi-grit/rapid-curation.
** Assembly quality assessment **
A Hi-C contact map was produced for the final version of the assembly. The Hi-C reads were aligned using bwa-mem2 ( Vasimuddin et al., 2019) and the alignment files were combined using SAMtools ( Danecek et al., 2021). The Hi-C alignments were converted into a contact map using BEDTools ( Quinlan & Hall, 2010) and the Cooler tool suite ( Abdennur & Mirny, 2020). The contact map is visualised in HiGlass ( Kerpedjiev et al., 2018). The Merqury.FK tool ( Rhie et al., 2020), run in a Singularity container ( Kurtzer et al., 2017), was used to evaluate the assembly QV scores. The genome was also analysed within the BlobToolKit environment ( Challis et al., 2020) and BUSCO scores ( Manni et al., 2021) were calculated. Table 4 contains a list of relevant software tool versions and sources.
** Taxonomic verification **
There are no published sequences of the Corticium candelabrum holotype (LMJG 15353/0) or other type material. For molecular taxonomic verification CO1 (Folmer) and 28S rDNA (C-Region) barcoding markers of the present sample were compared against sequences currently published in NCBI Genbank as C. candelabrum using MAFFT ( Katoh & Standley, 2013). The CO1 marker of present sample is identical to C. candelabrum JX999073 of Riesgo et al. (2014). However, alignments displayed only 94.5 % similarity (CO1) and 78% similarity (28S) to a C. candelabrum specimen published by Gazave et al. (2010) (HQ269363 and HM118553 respectively). As the latter specimen was collected in a cave environment (Coral Cave, Marseilles, France, see Gazave et al. (2010), which is not mentioned in Schmidt's original description of C. candelabrum (1862), we assume it being a different species.
Genome annotation
The Ensembl Genebuild annotation system ( Aken et al., 2016) was used to generate annotation for the Corticium candelabrum assembly (GCA_963422355.1) in Ensembl Rapid Release at the EBI. Annotation was created primarily through alignment of transcriptomic data to the genome, with gap filling via protein-to-genome alignments of a select set of proteins from UniProt ( UniProt Consortium, 2019).
Metagenome assembly
The metagenome assembly was generated using MetaMDBG ( Benoit et al., 2024) and binned using MetaBAT2 ( Kang et al., 2019), MaxBin ( Wu et al., 2014), bin3C ( DeMaere & Darling, 2019), and MetaTOR. The resulting bin sets of each binning algorithm were optimised and refined using MAGScoT ( Rühlemann et al., 2022). PROKKA ( Seemann, 2014) was used to identify tRNAs and rRNAs in each bin, CheckM ( Parks et al., 2015) (checkM_DB release 2015-01-16) was used to assess bin completeness/contamination, and GTDB-TK ( Chaumeil et al., 2022) (GTDB release 214) was used to taxonomically classify bins. Taxonomic replicate bins were identified using dRep ( Olm et al., 2017) with default settings (95% ANI threshold). The final bin set was filtered for bacteria and archaea. All bins were assessed for quality and categorised as metagenome-assembled genomes (MAGs) if they met the following criteria: contamination ≤ 5%, presence of 5S, 16S, and 23S rRNA genes, at least 18 unique tRNAs, and either ≥ 90% completeness or ≥ 50% completeness with fully circularised chromosomes. Bins that did not meet these thresholds, or were identified as taxonomic replicates of MAGs, were retained as ‘binned metagenomes’ provided they had ≥ 50% completeness and ≤ 10% contamination. A cladogram based on NCBI taxonomic assignments was generated using the ‘taxonomizr’ package in R. The tree was visualised and annotated using iTOL ( Letunic & Bork, 2024). Software tool versions and sources are given in Table 4.
Wellcome Sanger Institute – Legal and Governance
The materials that have contributed to this genome note have been supplied by a Tree of Life collaborator. The Wellcome Sanger Institute employs a process whereby due diligence is carried out proportionate to the nature of the materials themselves, and the circumstances under which they have been/are to be collected and provided for use. The purpose of this is to address and mitigate any potential legal and/or ethical implications of receipt and use of the materials as part of the research project, and to ensure that in doing so we align with best practice wherever possible. The overarching areas of consideration are:
• Ethical review of provenance and sourcing of the material
• Legality of collection, transfer and use (national and international)
Each transfer of samples is undertaken according to a Research Collaboration Agreement or Material Transfer Agreement entered into by the Tree of Life collaborator, Genome Research Limited (operating as the Wellcome Sanger Institute) and in some circumstances other Tree of Life collaborators.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdennur N Mirny LA : Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36(1):311–316. 10.1093/bioinformatics/btz 540 31290943 PMC 8205516 · doi ↗ · pubmed ↗
- 2Aken BL Ayling S Barrell D : The Ensembl gene annotation system. Database (Oxford). 2016;2016: baw 093. 10.1093/database/baw 093 27337980 PMC 4919035 · doi ↗ · pubmed ↗
- 3Allio R Schomaker-Bastos A Romiguier J : Mito Finder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020;20(4):892–905. 10.1111/1755-0998.13160 32243090 PMC 7497042 · doi ↗ · pubmed ↗
- 4Benoit G Raguideau S James R : High-quality metagenome assembly from long accurate reads with meta MDBG. Nat Biotechnol. 2024;42(9):1378–1383. 10.1038/s 41587-023-01983-6 38168989 PMC 11392814 · doi ↗ · pubmed ↗
- 5Boute N Exposito J Boury‐Esnault N : Type IV collagen in sponges, the missing link in basement membrane ubiquity*. Biol Cell. 1996;88(1–2):37–44. 10.1016/s 0248-4900(97)86829-3 9175266 · doi ↗ · pubmed ↗
- 6Challis R Richards E Rajan J : Blob Tool Kit – interactive quality assessment of genome assemblies. G 3 (Bethesda). 2020;10(4):1361–1374. 10.1534/g 3.119.400908 32071071 PMC 7144090 · doi ↗ · pubmed ↗
- 7Chaumeil PA Mussig AJ Hugenholtz P : GTDB-Tk v 2: memory friendly classification with the genome taxonomy database. Bioinformatics. 2022;38(23):5315–5316. 10.1093/bioinformatics/btac 672 36218463 PMC 9710552 · doi ↗ · pubmed ↗
- 8Cheng H Concepcion GT Feng X : Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18(2):170–175. 10.1038/s 41592-020-01056-5 33526886 PMC 7961889 · doi ↗ · pubmed ↗