Identifying prognostic targets in metastatic prostate cancer beyond AR
Emily Feng, Eric Feng, Tracy Berg, Isabella S. Nguyen, Lilac G. Nguyen, William Chen, Meng Zhang, David Quigley, Marina Sharifi, Haolong Li, Ilsa Coleman, Peter S. Nelson, Martin Sjöström, Shuang G. Zhao

TL;DR
The study identifies eight genes that could serve as new therapeutic targets for metastatic prostate cancer based on their strong cell line dependency and poor clinical outcomes.
Contribution
Combining functional screens with gene expression and clinical data to discover new prostate cancer drug targets beyond the androgen receptor.
Findings
Eight genes (CYC, CYP51A1, DHFR, EBP, KIF15, PPM1D, SQLE, and UMPS) showed strong cell line dependency and worse clinical outcomes.
Four of these genes (DHFR, EBP, KIF15, and PPM1D) are more highly expressed in neuroendocrine prostate cancer.
Most of these genes remain targetable post-abiraterone therapy as they are not significantly decreased post-treatment.
Abstract
Genome‐wide screens using CRISPR/RNAi can identify new therapeutic vulnerabilities in prostate cancer. In this study, we combine DepMap functional screen data with a large gene expression database (N = 1012) and clinical outcomes to identify potentially druggable targets. Eight genes (CYC, CYP51A1, DHFR, EBP, KIF15, PPM1D, SQLE, and UMPS) demonstrated strong dependency in cell lines and were also associated with worse prognosis clinically, representing potential therapeutic targets in metastatic prostate cancer. Four of these (DHFR, EBP, KIF15, and PPM1D) demonstrated higher expression in neuroendocrine prostate cancer. Furthermore, all but one (KIF15) were not significantly decreased from pretreatment to posttreatment, suggesting that they may remain targetable postabiraterone therapy. All eight genes showed evidence of protein expression in prostate cancers or cell lines. These…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
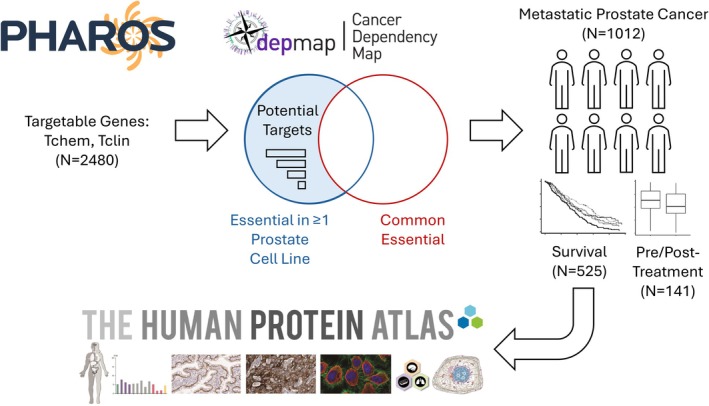 Fig. 1
Fig. 1 Fig. 2
Fig. 2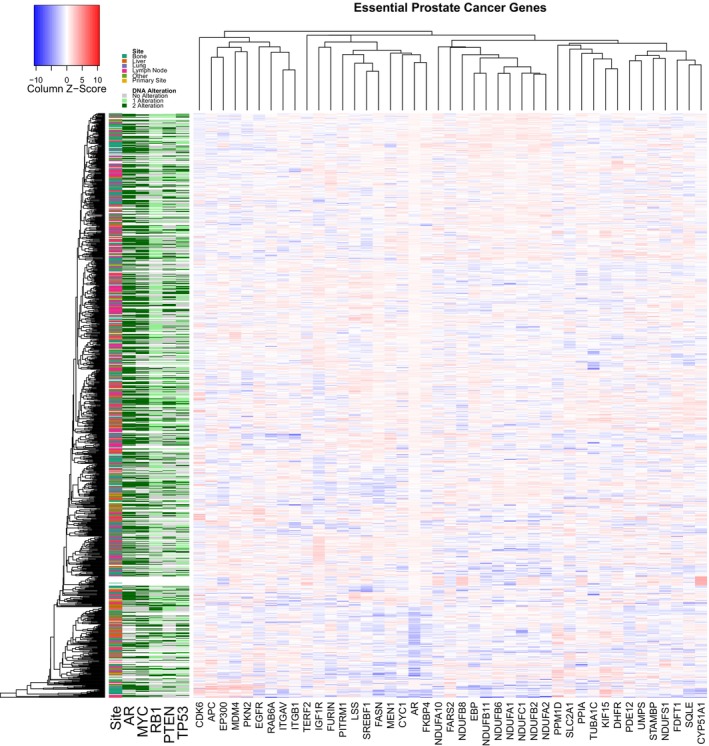 Fig. 3
Fig. 3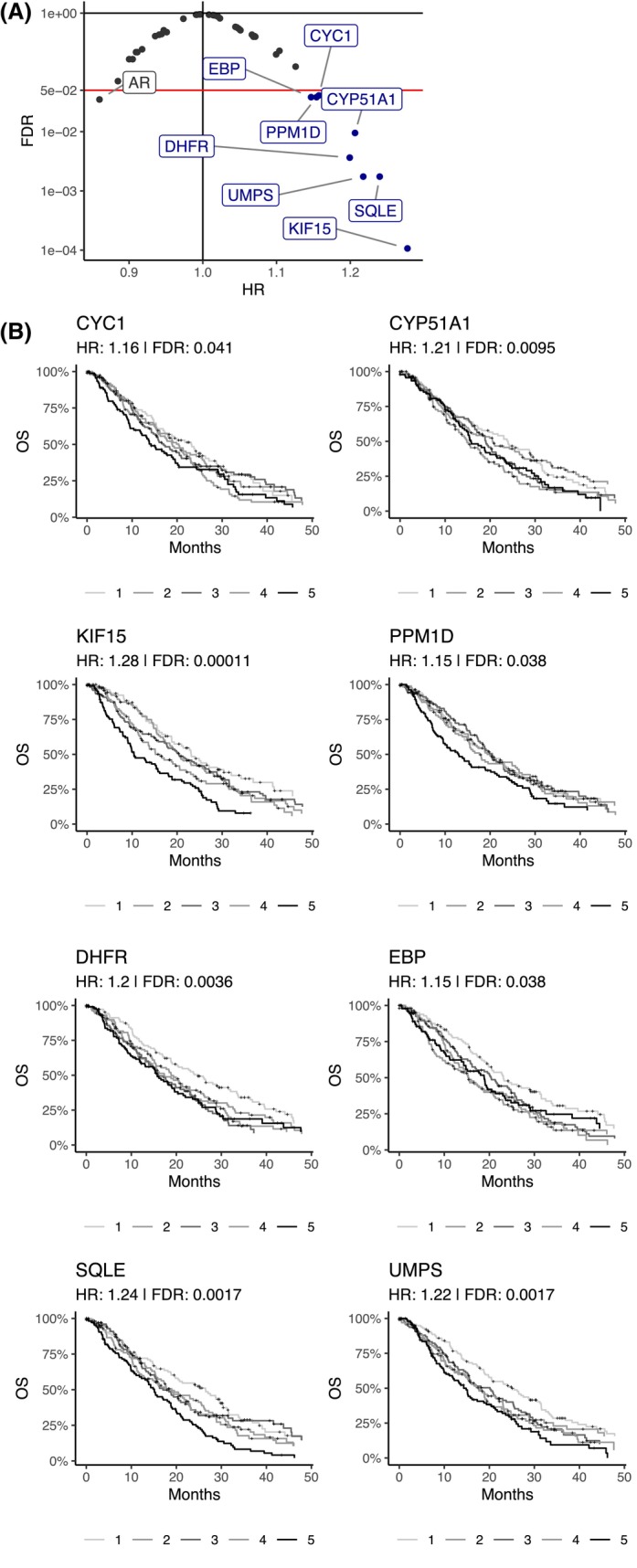 Fig. 4
Fig. 4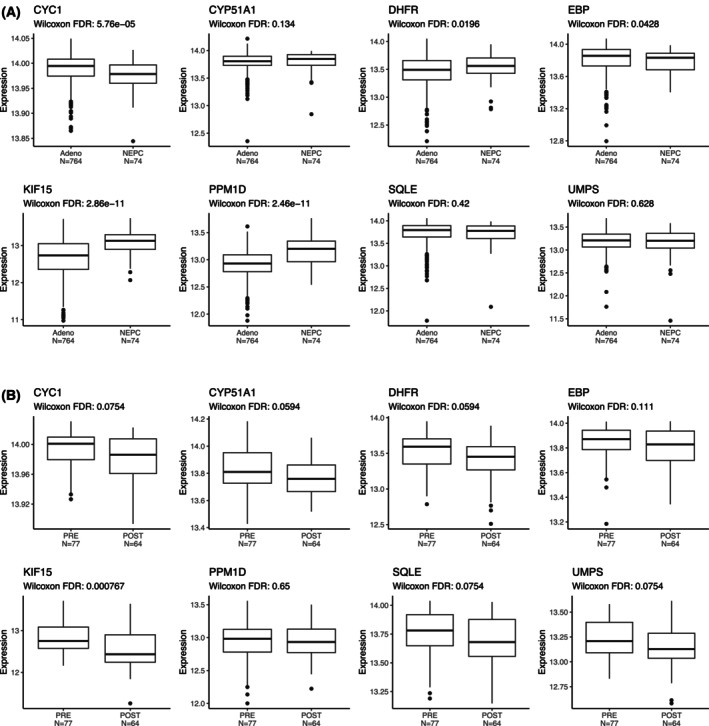 Fig. 5
Fig. 5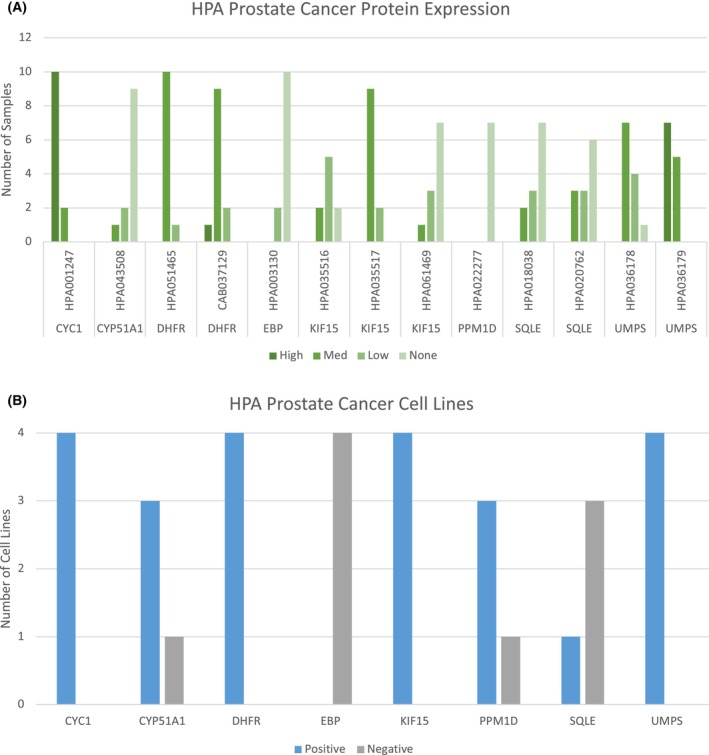 Fig. 6
Fig. 6- —NIH10.13039/100000002
- —DoD10.13039/100000005
- —Institute for Prostate Cancer Research10.13039/100017453
- —Prostate Cancer Foundation10.13039/100000892
- —Swedish Cancer Society (Cancerfonden)
- —Swedish Prostate Cancer Foundation (Prostatacancerförbundet)
- —Hjelms Stiftelse för Medicinsk Forskning
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProstate Cancer Treatment and Research · Prostate Cancer Diagnosis and Treatment · Cancer, Lipids, and Metabolism
High‐throughput methods for identifying novel therapeutic targets in cancer have been widely sought. Early work by the Cancer Cell Line Encyclopedia (CCLE) focused on screening drugs across hundreds of cancer cell lines [1]. However, drug screens cannot identify targets for which there is no compound yet. Large‐scale pooled functional genetic screens represent a way to simultaneously perturb thousands of genes and assess the effect on cell fitness. While CRISPR screens have been performed using various approaches in prostate cancer [2, 3, 4, 5, 6, 7], it is difficult to generalize their results given inconsistent methodology and small numbers of cell lines used. The Cancer Dependency Map (DepMap) has provided a more standardized and large‐scale screening of hundreds of cancer cell lines using both genome‐wide RNAi screens [8, 9], and subsequently CRISPR‐Cas9 (which will be abbreviated to just CRISPR subsequently) screens [10].
Prostate cancer is the most common malignancy in men according to the CDC US Cancer Statistics, with metastatic disease still lethal despite numerous FDA‐approved therapies. Utilization of functional screens [2, 3, 4, 5, 6, 7, 8, 9, 10] alone to identify prostate cancer dependencies and targets faces several challenges. Prostate cancer has only 10 total cell lines represented in DepMap [8, 9, 10], with an even smaller subset that is widely used, compared to other common cancer types with dozens of suitable model systems to represent the heterogeneity observed clinically. Therefore, it is critical to complement DepMap screen data [8, 9, 10] with other clinical data, especially in metastatic prostate cancer [11].
We assembled a dataset of 1012 metastatic prostate tumor samples with gene expression profiling [11]. This dataset was updated with additional overall survival (OS) data [12], to a new total of 525 patients, representing the largest aggregated clinical dataset in metastatic prostate cancer to our knowledge. Using these datasets, we first identified potentially druggable [13] targets based on in vitro genome‐wide functional screens [8, 9, 10]. We then examined clinical correlations and prioritized potential therapeutic targets in metastatic prostate cancer.
Materials and Methods
DepMap
DepMap data were downloaded from depmap.org. To do so, a model context was created with the following condition: Lineage is Prostate. Then, a custom download was initiated using this context as well as ‘Exclude columns and rows of NA's from download files’ and ‘Add cell line metadata to download’. CRISPR (DepMap Public 24Q2 + Score, Chronos), RNAi (Achilles + DRIVE + Marcotte, DEMETER2), and Batch corrected Expression Public 24Q4 data were then downloaded.
Metastatic prostate cancer datasets
Gene expression data in metastatic prostate cancer were obtained from five published datasets: the University of Washington/Fred Hutchinson Cancer Center (UW/FHCRC) autopsy cohort (n = 254) [14, 15], a neuroendocrine prostate cancer (NEPC)‐enriched dataset from Weill Cornell Medicine (WCM) (n = 49) [16], the Castration Resistant Prostate Cancer (CRPC) datasets from the Stand Up 2 Cancer/Prostate Cancer Foundation (SU2C/PCF) East Coast Dream Team (ECDT; n = 328) [12, 17, 18], and West Coast Dream Team (WCDT; n = 240) [19, 20, 21, 22, 23, 24, 25] datasets. The Prostate Cancer Medically Optimized Genome‐Enhanced Therapy (PROMOTE) trial (n = 141) [26] contained pre‐ and postabiraterone exposure samples. Overall survival data were available for a subset of samples from the WCDT, ECDT, and PROMOTE cohorts (n = 525). Full details of these datasets are extensively described in their original publications [14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26]. DNA alteration calls were available for 780 samples, and adenocarcinoma versus NEPC status was available for 838 samples [11], as defined in each cohort.
Bioinformatics analysis
We also limited our analysis to genes that are potentially targetable by existing molecules or clinical drugs (Tchem, Tclin respectively) according to Pharos (the web interface for the NIH Illuminating the Druggable Genome program) [27]. CRISPR screen data were used for prostate cancer and transformed prostate epithelial cell lines, but only RNAi screen data were available for NEPC. CRISPR and RNAi screen analysis was performed using DepMap [9]. Per the DepMap FAQ, a gene effect < −1 in CRISPR/RNAi screens is the median of all pan‐essential genes and a threshold that indicates strong killing. We identified genes where at least one prostate cancer/transformed cell line met this threshold in a cell line in which the gene was expressed, indicating some effect in at least a subset. Normalization of the gene expression data from the metastatic prostate cancer datasets was performed as previously described [11, 28, 29]. Pathway enrichment analysis was performed using the NIH david tool [30]. Protein expression was confirmed in the Human Protein Altas (HPA; www.proteinatlas.org).
Statistical analysis
Survival analysis was performed using Cox regression with gene expression as a continuous variable, and overall survival as the endpoint in our samples with clinical outcomes. Multiple testing correction was performed using the Benjamini–Hochberg method. We also performed comparisons of expression between adenocarcinoma vs. NEPC and pre‐ vs. postabiraterone, and used a Wilcoxon signed‐rank test. All analysis was completed in r version 4.2.2 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Overview
Prior studies indicate that 2480 genes are known to encode for proteins that bind to small molecules (PHAROS: Tchem), with some targeted by known drugs (PHAROS: Tclin). We queried DepMap CRISPR screens in prostate cancer cell lines as well as transformed prostate epithelial cell lines for evidence that these genes may alter cellular fitness. A gene effect < 0 indicates a deleterious effect, which can be observed for AR in VCaP (−0.333), LNCaP (−0.78), and 22RV1 (−1.28), all known to be androgen‐dependent [31]. On the other hand, known androgen‐independent cell lines [32, 33] (e.g., DU145, PC3) all had AR gene effects > 0. We therefore deemed genes where disruption caused a gene effect < −1 (the median of all pan‐essential genes indicating strong essentiality) as the most essential. Given the heterogeneity of prostate cancer and the small number of representative cell lines, we retained genes that were essential in one or more cell lines. NCIH660, the only neuroendocrine prostate cancer (NEPC) cell line, did not have CRISPR screen data available, but did have RNAi screen data, and so we utilized the RNAi screen data for this cell line in order to include NEPC, with the same thresholds. We also removed any common essential genes (per DepMap; Fig. 1).
Schematic of this study. We limited our analysis to genes that are potentially targetable by existing molecules or clinical drugs (Tchem, Tclin respectively) according to Pharos (the web interface for the NIH Illuminating the Druggable Genome program). CRISPR/RNAi screen data from The Cancer Dependency Map (DepMap) were then used to identify targets that were essential for prostate cancer and transformed prostate epithelial cell lines but that were not commonly essential. Per the DepMap frequently asked questions (FAQ), a gene effect < −1 in CRISPR/RNAi screens is the median of all pan‐essential genes and a threshold that indicates strong killing. We identified genes where at least one prostate cancer/transformed cell line met this threshold, indicating some effect in at least a subset. We then identified genes where expression was associated with clinical outcomes from published datasets, resulting in nine genes including AR. Protein expression in the tumor was confirmed in the Human Protein Altas.
Functional screening reveals prostate cancer‐specific dependencies
The integrated analysis of all prostate cancer cell line models resulted in 43 total genes with strong effects in prostate cancer/transformed cell lines from these functional screens (Fig. 2). Reassuringly, AR was identified in this list of potential therapeutic targets. Only one gene, KIF15, demonstrated a strong effect from disruption in the NEPC cell line. Using david pathway enrichment analysis [30], the prostate cancer dependencies were enriched for genes related to metabolism, and particularly cholesterol metabolism (Table S1). We next examined these targets in a previously assembled clinicogenomic metastatic prostate cancer dataset with 1012 metastatic prostate cancer samples [11]. In general, the gene expression did not show strong overall correlations with each other, biopsy site, or the most common genomic alterations in metastatic prostate cancer such as AR mutations/amplifications, MYC amplification, or RB1/TP53/PTEN loss (Fig. 3).
Essential prostate cancer gene dependencies. This figure shows the gene effects from The Cancer Dependency Map (DepMap) CRISPR/RNAi screens for all genes that are essential in at least one prostate cancer or transformed prostate cell line. This was defined as a gene effect of −1 (the median of all pan‐essential genes, and a threshold that indicates strong killing per the DepMap frequently asked questions (FAQ)) and indicated with the purple vertical line. The light/medium/dark blue vertical lines indicate the gene effect of AR CRISPR in all prostate cancer cell lines with a deleterious gene effect in the DepMap (−0.333: VCaP, −0.78: LNCaP, −1.28 in 22RV1).
Expression of essential prostate cancer genes. Heatmap of the gene expression of the essential prostate cancer genes (in one or more cell lines) from Fig. 2, as well as the site of biopsy and key oncogenic and tumor suppressor alterations.
Clinical outcomes associations
We next examined clinical outcomes by examining associations between genes with fitness effects in vitro and OS in the 525 samples where these data were available, the largest such dataset to our knowledge. Using continuous Cox regression, we found that 12 of the prostate cancer dependencies had statistically significant P‐values, which decreased to 9 after the Benjamini–Hochberg FDR correction was applied. Eight of these were associated with a Hazard Ratio (HR) > 1 indicating worse prognosis with increasing expression (Fig. 4). AR was the only gene where higher expression was associated with a better prognosis (compared to lower expression), which is concordant with the decrease in AR signaling in aggressive basal and NEPC tumors and the decreased efficacy of agents targeting AR [19, 28, 29]. The eight genes where higher expression was associated with worse prognosis (CYC, CYP51A1, DHFR, EBP, KIF15, PPM1D, SQLE, and UMPS) represent potential additional therapeutic targets in metastatic prostate cancer.
Clinical outcomes of essential prostate cancer genes. (A) Volcano plot of continuous gene expression Cox regression for Overall Survival (OS) of False Discover Rates (FDRs) and Hazard Ratios (HRs, scaled by standard deviation) of the essential prostate cancer genes (in one or more cell lines) from Figs 2 and 3. (B) Kaplan–Meier curves of the eight genes with an FDR < 0.05 and HR > 1, split into quintiles. The continuous HRs and FDRs are shown.
NEPC, pre‐/postabiraterone
NEPC is an aggressive, androgen‐independent subtype of prostate cancer [19, 28, 29]. We next determined whether these eight targets were differentially expressed in metastatic NEPC tumors compared to metastatic prostate adenocarcinomas. DHFR, EBP, KIF15, and PPM1D demonstrated higher expression in NEPC, whereas CYC1 demonstrated lower expression (Fig. 5A). We also took advantage of the pretreatment samples before abiraterone therapy and those taken after 12 weeks of abiraterone from the PROMOTE trial to examine whether treatment with abiraterone modulated gene expression. Only KIF15 was significantly, though only modestly, decreased from pretreatment to posttreatment (Fig. 5B), suggesting that these genes may remain targetable postabiraterone therapy.
Histology, pre/post‐abiraterone expression of essential prostate cancer genes. Boxplots and Wilcoxon Rank‐Sum P‐values of expression (log‐transformed normalized gene rank) comparing (A) Adenocarcinoma vs. neuroendocrine prostate cancer (NEPC; as defined in the original publications of the cohorts) and (B) pre‐ vs. 12 weeks postabiraterone initiation samples in the Prostate Cancer Medically Optimized Genome‐Enhanced Therapy (PROMOTE) trial of the eight prognostic and essential prostate cancer genes (in one or more cell lines) from Fig. 4B.
Protein expression
Six of these eight genes demonstrated medium–high protein expression in one or more prostate tumors in the HPA (Fig. 6A). EBP did not show medium–high protein expression but did demonstrate low expression in two prostate tumors. While PPM1D did not demonstrate expression in clinical prostate tumors, it did show protein expression in 75% (3/4) prostate cancer cell lines tested (Fig. 6B). Altogether, all eight targets showed some evidence of protein expression in prostate cancers or cell lines. The HPA validates each of the antibodies for IHC and assigns them one of four classifications: Uncertain, Approved, Supported, and Enhanced. The antibodies for six of the eight targets were all Approved, Supported, or Enhanced. The antibodies used for DHFR and UMPS were classified as Uncertain. However, each of these proteins had two different antibodies each, providing orthogonal validation of protein expression in at least some prostate tumors.
Human protein Atlas confirmation of tumor protein expression. (A) Barplots of Human Protein Atlas (HPA) expression in prostate cancer samples and (B) Barplots of HPA expression in prostate cancer cell lines (22Rv1, NCI‐H660, PC‐3, VCaP) of the eight genes from Figs 4B and 5. Positive/Negative defined per HPA based on detected/not detected using Mass Spectrometry.
Discussion
Herein, we performed a clinicogenomic evaluation of genes conferring cell fitness defects in vitro and identified eight potential therapeutic targets in metastatic prostate cancer. Inactivation of these targets created strong phenotypes in one or more prostate cancer/transformed cell lines. Furthermore, increased gene expression of these targets was associated with worse prognosis in a large clinical cohort. There is also support for prostate cancer tissue or cell line protein expression of all eight targets, to varying degrees.
These eight genes have support as drug targets in prostate and other cancers. Notably, three genes involved in cholesterol synthesis pathways were identified, CYP51A1, SQLE, and EBP. Cholesterol synthesis is required by cancer cells to form membranes and supply energy for their rapid growth and division [34], which has led to exploration of its inhibition across multiple cancer types [35]. Of particular importance to prostate cancer, androgens are also synthesized from cholesterol precursors [36]. Androgen biosynthesis can play a role in prostate cancer progression and is a target of multiple drugs used to treat advanced disease: abiraterone [37] and ketaconazole [38]. While the primary target of these drugs is CYP17A1, both have also been shown to potentially target CYP51A1 [39, 40]. SQLE is upregulated across multiple cancers [41], both at the level of gene duplication and protein stabilization after p53/PTEN downregulation [35]. Elevated SQLE has also been associated with high‐risk prostate cancer, and inhibition promotes prostate cancer cell death [42, 43]. Its inhibition has also been shown to cause toxic accumulation of squalene in neuroendocrine lung cancer cells [44]. Interestingly, both CYP51A1 and SQLE (squalene epoxidase) gene expression may be involved in suppressing the ferroptosis cell death pathway [45, 46]. Ferroptosis is triggered by accumulation of iron and lipid peroxidases, and inducers of ferroptosis have been proposed as promising cancer drugs in prostate and other cancers [47]. EBP (emopamil‐binding protein) encodes cholestenol delta‐isomerase, an endoplasmic reticulum membrane protein also involved in cholesterol biosynthesis that also likely binds and transports multiple drugs [48]. Inhibition of EBP has been shown to inhibit prostate cancer cell proliferation [49, 50].
We also identified genes needed for nucleotide synthesis, required for rapidly replicating cells. DHFR encodes the dihydrofolate reductase protein essential for synthesis of purines and thymidylate [51]. It is the target of the cancer therapeutic methotrexate as well as several newer drugs in clinical use and in development [51]. Methotrexate has been investigated in small trials in prostate cancer [52, 53], but only in combination with other chemotherapies. UMPS encodes uridine 5'‐monophosphate synthase, a bifunctional enzyme [54] involved in de novo synthesis of pyrimidines. Inhibitors of this and other steps of de novo pyrimidine synthesis are under active investigation in cancer [54, 55, 56, 57]. The pathway may make a promising drug target, as rapid proliferation of cancer cells depletes substrates from salvage pyrimidine biosynthesis pathways, increasing dependence on the de novo pathway [57]. Interestingly, this pathway has also been linked to cell defense against ferroptosis [54].
Other genes were identified which affect metabolic pathways, protein transport, and apoptosis. The CYC1 gene encodes cytochrome C1, a mitochondrial protein that is part of the electron transport chain driving oxidative phosphorylation, which may play a role in metabolic plasticity that promotes stemness, drug resistance, and metastasis [58]. This metabolic plasticity is increasingly recognized in prostate cancer [58], though the role of CYC1 specifically in this process has not been explored. However, elevated CYC1 with a potential function in resisting apoptosis has been found in uveal melanoma [59] and osteosarcoma [60, 61]. Kinesin family member 15 (KIF15) is overexpressed in multiple cancers and is involved in mitotic spindle assembly and protein transport [62]. Inhibitors of KIF15 are under development and have been demonstrated to prevent mitosis preclinically in combination with other drugs [63]. Its role in multiple cancers is currently being explored [64, 65, 66, 67], including prostate cancer, in which it may play a role in resistance to AR inhibition [68, 69]. The PPM1D gene encodes protein phosphatase, Mg2+/Mn2+‐dependent 1D, a Ser/Thr protein phosphatase also known as wild‐type p53‐induced phosphatase 1 (Wip1) and is a negative regulator of p53‐induced apoptosis. Germline mutations in this gene and overexpression are associated with increased risks of multiple cancers [70], including prostate cancer [70, 71, 72] and many hematological malignancies [73].
High‐throughput CRISPR and RNAi screens have several limitations, and validation of anticancer activity can depend on the model system, method of inhibition, and other factors. Even in the setting of known driver alterations such as the TMPRSS2‐ERG (T2E) fusion, there can be inconsistent effects. VCAP contains a T2E fusion and is also highly dependent on ERG, with a gene effect of −1.43 for ERG in the DepMap CRISPR screen. NCIH660 also has a T2E fusion but with no gene effect for ERG in the DepMap CRISPR screen, which may be because NCIH660 is an NEPC model and thus likely has other alterations driving this transformation that make it less dependent on the T2E fusion. Nonetheless, these screens identify AR as a key target in prostate cancer. The other eight targets are associated with worse clinical outcomes and demonstrate varying levels of literature support as potential targets, supporting further preclinical and clinical investigations. However, it is important to note that these targets are only essential in a subset of cell lines. Given the differences molecularly between the cell lines, further investigation into potential interactions with specific DNA alterations is important to better understand the potentially targetable patient subgroups. Furthermore, additional protein studies will be important, given that HPA focuses on localized disease and the antibodies used have varying levels of validation.
Conflict of interest
SGZ has patent applications with Veracyte on molecular signatures in prostate cancer unrelated to this work, and a family member employed by Artera, and with stock in Exact Sciences. MSj reports speaker fees from Astellas and consulting fees and Advisory Board for Astellas/Adelphi Targis, unrelated to this work. PSN has served as a paid consultant to Janssen, Genentech, Pfizer, and AstraZeneca and received research support from Janssen for work unrelated to the present report.
Author contributions
MS and SGZ conceived and designed the project. WC, MZ, DQ, MS, HL, IC, PSN, MS, and SGZ acquired the data. EF, EF, ISN, LGN, WC, MZ, DQ, MS, HL, PSN, MS, and SGZ analyzed and interpreted the data. TB, MS, and SGZ wrote the paper.
Supporting information
Table S1. Pathway analysis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Barretina J , Caponigro G , Stransky N , Venkatesan K , Margolin AA , Kim S , Wilson CJ , Lehár J , Kryukov GV , Sonkin D et al. (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607.22460905 10.1038/nature 11003 PMC 3320027 · doi ↗ · pubmed ↗
- 2Ahmed M , Soares F , Xia J‐H , Yang Y , Li J , Guo H , Su P , Tian Y , Lee HJ , Wang M et al. (2021) CRISP Ri screens reveal a DNA methylation‐mediated 3D genome dependent causal mechanism in prostate cancer. Nat Commun 12, 1781.33741908 10.1038/s 41467-021-21867-0PMC 7979745 · doi ↗ · pubmed ↗
- 3Das R , Sjöström M , Shrestha R , Yogodzinski C , Egusa EA , Chesner LN , Chen WS , Chou J , Dang DK , Swinderman JT et al. (2021) An integrated functional and clinical genomics approach reveals genes driving aggressive metastatic prostate cancer. Nat Commun 12, 4601.34326322 10.1038/s 41467-021-24919-7PMC 8322386 · doi ↗ · pubmed ↗
- 4Rodriguez Y , Unno K , Mihai T I , Chalmers ZR , Yoo YA , Vatapalli R , Sagar V , Yu J , Lysy B , Hussain M et al. (2022) A genome‐wide CRISPR activation screen identifies PRRX 2 as a regulator of enzalutamide resistance in prostate cancer. Cancer Res 82, 2110–2123.35405009 10.1158/0008-5472.CAN-21-3565 PMC 9177667 · doi ↗ · pubmed ↗
- 5Tang S , Sethunath V , Metaferia N , Nogueira M , Gallant D , Garner E , Lairson L , Penney C , Li J , Gelbard M et al. (2022) A genome‐scale CRISPR screen reveals PRMT 1 as a critical regulator of androgen receptor signaling in prostate cancer. Cell Rep 38, 110417.35196489 10.1016/j.celrep.2022.110417 PMC 9036938 · doi ↗ · pubmed ↗
- 6Tsujino T , Takai T , Hinohara K , Gui F , Tsutsumi T , Bai X , Miao C , Feng C , Gui B , Sztupinszki Z et al. (2023) CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. Nat Commun 14, 252.36650183 10.1038/s 41467-023-35880-y PMC 9845315 · doi ↗ · pubmed ↗
- 7Fei T , Chen Y , Xiao T , Li W , Cato L , Zhang P , Cotter M , Bowden M , Lis R , Zhao S et al. (2017) Genome‐wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc Natl Acad Sci U S A 114, E 5207–E 5215.28611215 10.1073/pnas.1617467114 PMC 5495225 · doi ↗ · pubmed ↗
- 8Mc Donald E , de Weck A , Schlabach MR , Billy E , Mavrakis KJ , Hoffman GR , Belur D , Castelletti D , Frias E , Gampa K et al. (2017) Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large‐scale, deep RN Ai screening. Cell 170, 577–592 e 510.28753431 10.1016/j.cell.2017.07.005 · doi ↗ · pubmed ↗