Genome-Wide Identification of Novel miRNAs and Infection-Related Proteins in Leishmania major via Comparative Analysis of the Protozoa, Vectors, and Mammalian Hosts
Tianyi Liu, Jinyang Qian, Yicheng Yan, Xi Zeng, Zhiyuan Yang

TL;DR
This study identifies new microRNAs and infection-related proteins in Leishmania major, offering potential targets for treating leishmaniasis.
Contribution
The study presents novel miRNAs and infection-related proteins in Leishmania major through comparative genomic analysis.
Findings
2963 conserved proteins were identified across L. major, its vector, and mammalian hosts.
27 infection-related proteins, including 24 protein kinases, were discovered as potential therapeutic targets.
29 novel miRNAs in L. major were identified, with seven absent in all mammalian species.
Abstract
Leishmania major is a unicellular protozoan that causes cutaneous leishmaniasis in mammals and is mainly transmitted by the sand fly Phlebotomus papatasi. However, the contribution of microRNAs (miRNAs) and protein-coding genes to its pathogenic mechanisms remains largely unexplored. In this study, we systematically analyzed miRNAs and protein-coding genes in L. major, its insect vector, and mammalian hosts. Comparative genomic analysis revealed 2963 conserved proteins shared among the three groups, highlighting a core set of proteins across protozoa, vectors, and hosts. Among mammals, human proteins exhibited the highest homology with L. major, while P. papatasi displayed the lowest proportion of homologs. Functional annotation of 94 hypothetical proteins identified 27 infection-related proteins, including 24 protein kinases and three tyrosine phosphatases, which may represent novel…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
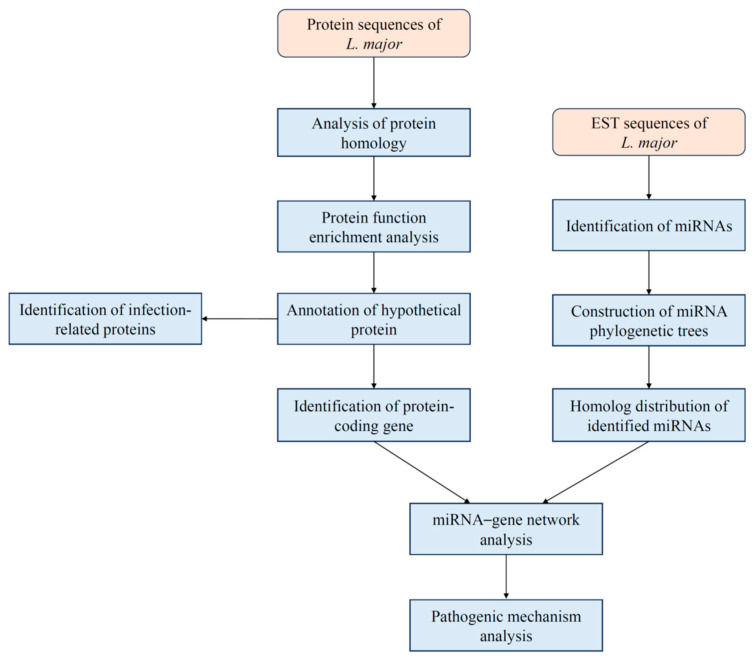 Figure 1
Figure 1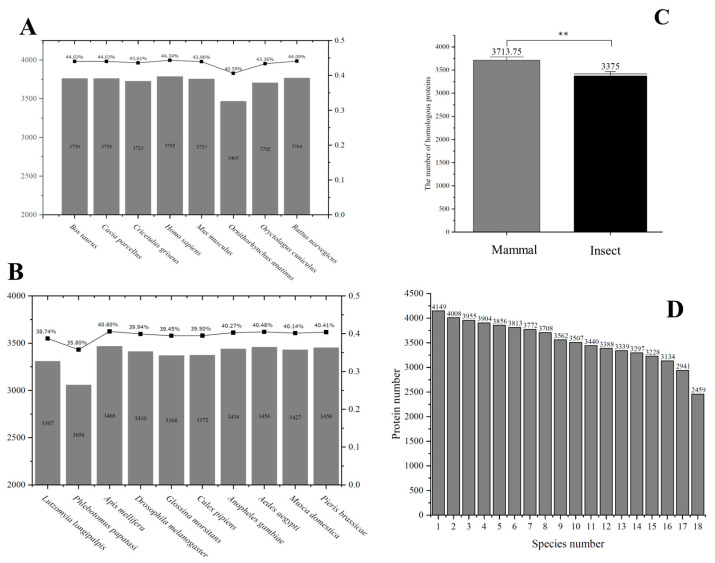 Figure 2
Figure 2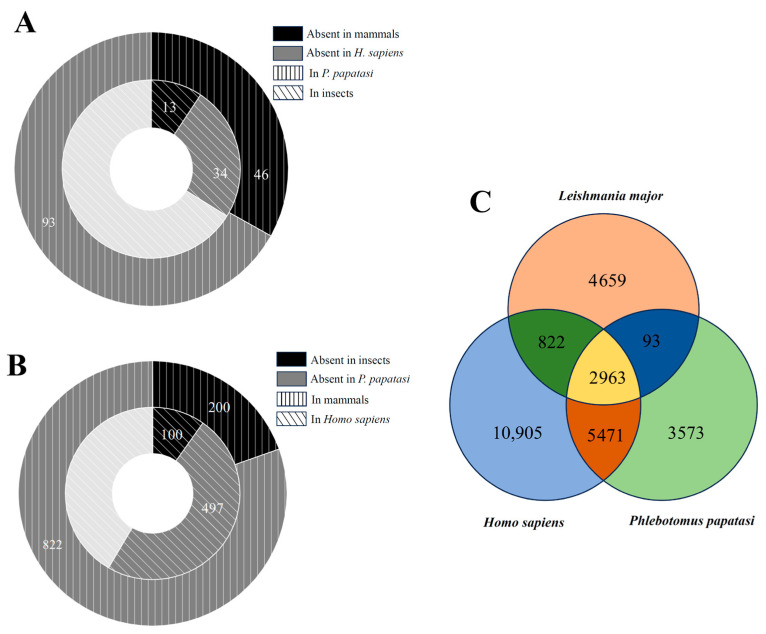 Figure 3
Figure 3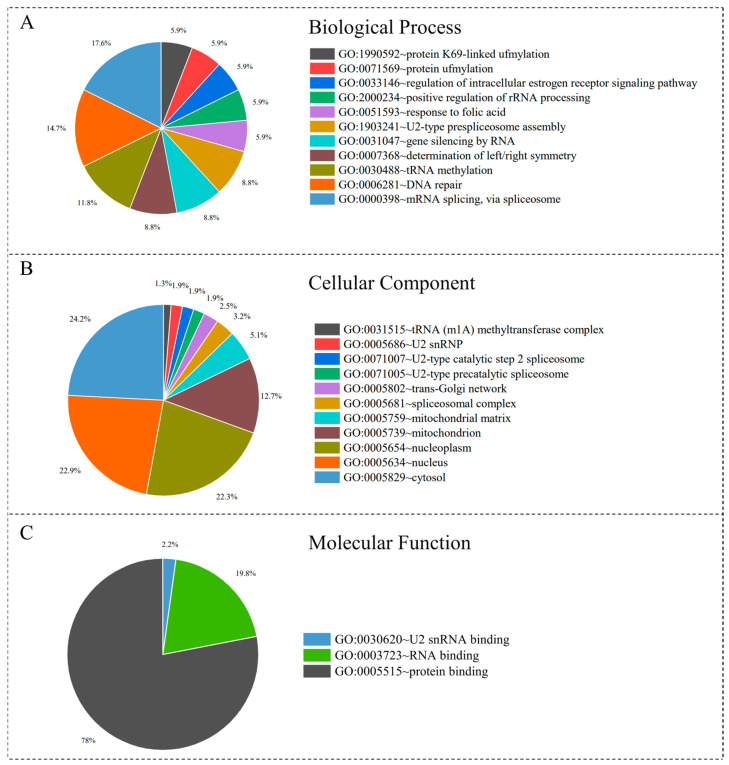 Figure 4
Figure 4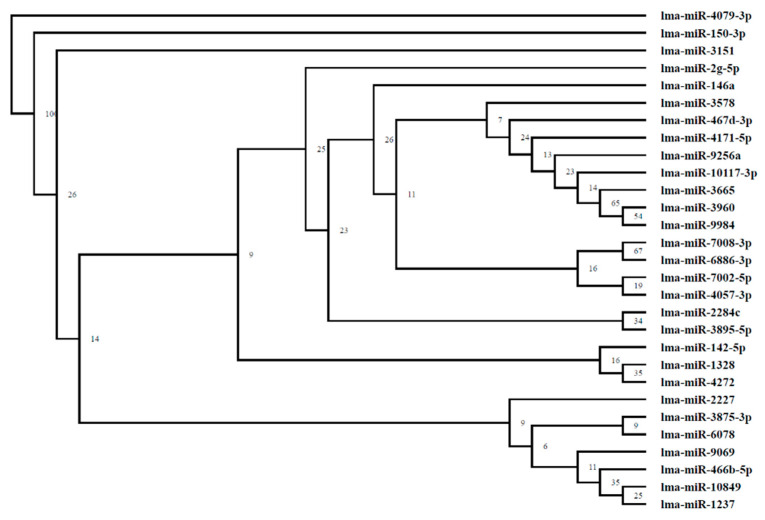 Figure 5
Figure 5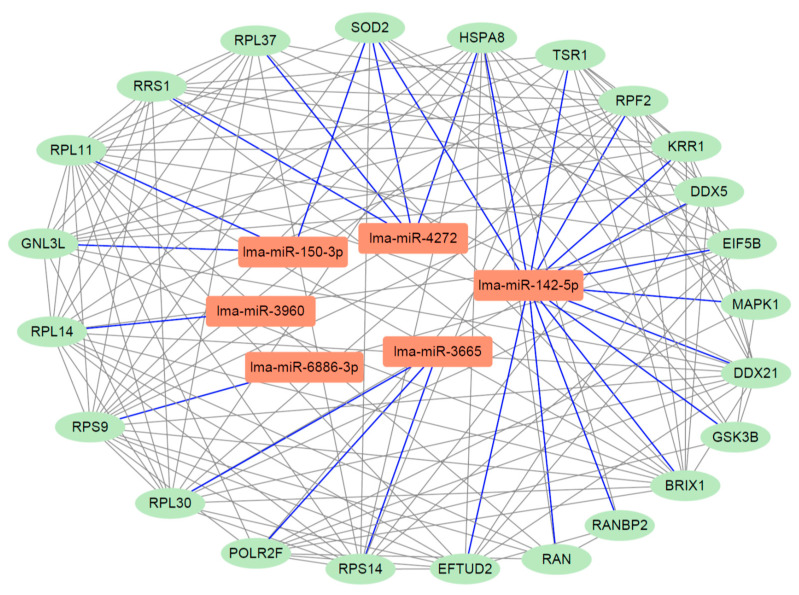 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsResearch on Leishmaniasis Studies · Trypanosoma species research and implications
1. Introduction
Leishmania major is a type of protozoan that causes cutaneous leishmaniasis, a vector-borne disease affecting mammals. Infections with L. major typically manifest as localized cutaneous lesions or ulcers at the site of the sand fly bite, characterized by swelling, inflammation, and tissue destruction [1]. Although extensively studied, important knowledge gaps remain, particularly regarding its pathogenic mechanisms and the development of effective interventions. Current control strategies mainly focus on reducing sand fly populations through vector control and promoting community education to minimize exposure [2]. However, no effective vaccine is yet available [3]. Thus, a deeper understanding of the biology and pathogenesis of L. major is essential for developing novel therapies, and protozoan control will continue to be a key approach for managing leishmaniasis in the future.
Genome annotation of L. major and the studies of its microRNAs (miRNAs) have become particularly important. The first annotated genome of L. major was published in 2005 [4] and updated in 2021 [5]. Despite these updates, the functions of many proteins remain unknown, underscoring the need for systematic re-annotation. Advances in bioinformatics have provided powerful tools for protein function prediction, which have been successfully applied to other pathogens such as bacterium Mycobacterium tuberculosis [6] and fungus Alternaria alternata [7]. These approaches hold promise for improving the annotation of L. major proteins.
In addition, relatively few studies have investigated the role of miRNAs in leishmaniasis. miRNAs are small non-coding RNAs, typically 18–24 nucleotides in length, that regulate protein expression at the post-transcriptional level. Evidence suggests that specific miRNAs play important roles in L. major infection, such as miR-146a [8]. With advances in computational approaches, novel miRNAs have been successfully identified from expressed sequence tags (ESTs) in species including plant Jatropha curcas [9] and protozoan Trypanosoma brucei [10].
The urgent need for novel therapeutic strategies against leishmaniasis is highlighted by the limitations of current treatments and the absence of a human vaccine. Recent approaches include combination therapies for cutaneous and visceral leishmaniases; drug repurposing strategies, such as with artesunate; and exploration of natural products with potential anti-parasitic activity. Protein kinases, in particular, have emerged as critical molecular targets for drug discovery in Leishmania, supported by genome sequencing data that provide insights into essential pathways for protozoan survival [11].
In this study, we applied a series of bioinformatics tools to identify protein-coding genes and miRNAs in L. major. We functionally annotated protozoan proteins with an emphasis on those related to virulence, and explored specific miRNAs potentially involved in infection. Collectively, our work provides a comprehensive overview of proteins, genes, and miRNAs in L. major, offering new insights into its pathogenic mechanisms and potential therapeutic targets. Given the clinical significance of leishmaniasis as a neglected tropical disease, our findings contribute to a better understanding of host–pathogen interactions.
2. Materials and Methods
2.1. Genome and Proteome Retrieval
The genome of Leishmania major, including 8537 protein-coding sequences and expressed sequence tag (EST) data, was retrieved from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/, NCBI). Proteomes of eight representative mammals and ten insects were obtained from the UniProt database (https://www.uniprot.org/) [12], while miRNA sequences for all animals were retrieved from the miRBase database (https://www.mirbase.org/) [13]. The overall workflow of our study is illustrated in Figure 1.
2.2. Protein Homology Analysis
Since L. major is transmitted by insects and infects mammalian hosts, we analyzed protein homology across three groups: the protozoa, mammals, and insects. Proteins from eight common mammalian species, including Bos taurus (bovine), Cavia porcellus (guinea pig), Cricetulus griseus (Chinese hamster), Homo sapiens (human), Mus musculus (mouse), Ornithorhynchus anatinus (platypus), Oryctolagus cuniculus (rabbit), and Rattus norvegicus (rat), were selected for comparison with L. major using BLAST (version 2.16) with an E-value cutoff of 1 × 10^−6^. This stringent threshold reduces the likelihood of false positives and thus increases the reliability of homologous alignments.
Similarly, ten insect species, including Aedes aegypti (yellow fever mosquito), Anopheles gambiae (African malaria mosquito), Apis mellifera (honey bee), Culex pipiens (common house mosquito), Drosophila melanogaster (fruit fly), Glossina morsitans (tsetse fly), Lutzomyia longipalpis (sand fly), Musca domestica (house fly), Phlebotomus papatasi (sand fly), and Pieris brassicae (large white butterfly), were compared against L. major proteins. The distribution of homologous proteins across the three groups was calculated and analyzed.
To assess uncertainty, we calculated standard errors and 95% confidence intervals (CI) as follows:
Here, SEM represents the standard error of the mean, SD is the sample standard deviation, N is the sample size, is the sample mean, and t is the t-value corresponding to a 95% confidence interval (α = 0.05) with N − 1 degrees of freedom.
2.3. Protein Function Enrichment Analysis
After removing duplicate alignments, we integrated the results and applied a bi-directional best hit (BBH) strategy [14] to identify the intersection set of proteins present in all mammalian species. Gene Ontology (GO) enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (https://davidbioinformatics.nih.gov/, DAVID) [15]. For each GO term, the number of associated proteins was counted, and terms with a p-value ≤ 0.05 were considered significantly enriched. Three GO categories—cellular component, molecular function, and biological process—were analyzed separately.
2.4. Hypothetical Protein Annotation
Due to incomplete annotation of the L. major genome, many proteins remain labeled as “hypothetical protein.” To improve annotation, these sequences were aligned against the non-redundant (nr) protein database in NCBI using BLAST. The top hits were extracted and used to assign putative functions to the hypothetical proteins in L. major.
2.5. Infection-Related Protein Identification
Protozoa rely on specific proteins to establish infection in mammalian hosts, making these proteins potential targets for therapeutic intervention. In L. major, key protein groups implicated in infection include tyrosine phosphatases, cytokines, and protein kinases. For example, L. major can secrete tyrosine phosphatases via exosomes to modulate host cell signaling [16], while cytokine gene expression can alter the function of infected human macrophages [17]. Protein kinases serve as critical regulators in the protozoan life cycle and are considered important molecular targets [18]. Based on these findings, we focused on these three protein groups in our analysis and discussion.
2.6. Identification of Novel miRNA
miRNA-mediated approaches have shown potential for future therapeutic applications in various diseases. Given the high conservation of miRNAs across species, novel miRNAs can be identified in target organisms through homology-based searches. Expressed sequence tags (ESTs) are short DNA fragments derived from complementary DNA, which can serve as markers for gene locations. The EST-based method has been widely applied for miRNA discovery in parasites [10] and was employed in this study. All known animal miRNAs were aligned with L. major EST sequences using BLAST (version 2.16). Candidate miRNAs were selected based on the following criteria:
- (1)Maximum of 2 mismatches allowed;
- (2)Prediction of the minimum free energy frequency (FMFE) of matching sequence fragments using RNAfold (version 2.3) [19], with a cutoff FMFE ≤ 0.05;
- (3)Prediction of hairpin-like secondary structures using RNAfold (version 2.3);
- (4)Significant negative values of minimum folding free energy (MFE) and MFE index (MFEI) for secondary structures, calculated using:
These thresholds are consistent with prior studies on RNA structure prediction, ensuring the reliability of our results.
2.7. Construction of miRNA Phylogenetic Tree
Phylogenetic analysis of miRNAs provides insights into their evolutionary relationships, helping to identify common ancestral miRNAs, infer potential shared functions, and determine conserved miRNAs across species. Multiple sequence alignment of the identified miRNAs was performed using Clustal Omega (version 1.2.2) [20] to detect common features. Based on the alignment, a phylogenetic tree was constructed using the maximum parsimony method, which seeks the simplest tree minimizing sequence changes. The reliability and stability of the tree were evaluated using a bootstrap-like self-unfolding approach, in which the tree was reconstructed multiple times and node support values were calculated. Nodes with higher support values were selected to construct the final phylogenetic tree, illustrating the evolutionary relationships among miRNAs.
2.8. Distribution of Identified miRNAs
Analyzing the distribution of homologous miRNAs can provide insights into their potential functions. The miRNAs identified in L. major were compared with homologous sequences in other species, and their distribution patterns were visualized. Given that mammals are the primary hosts of L. major, all known mammalian miRNAs were included in the comparison. miRNAs unique to L. major were selected for further functional analysis.
2.9. Construction of miRNA-Gene Interaction Network
Target genes of the identified miRNAs were predicted using miRWalk [21] and miRTarBase [22]. Based on these predictions, a miRNA–gene interaction network was constructed and visualized using Cytoscape (version 3.9.1) [23]. To identify key functional nodes within the network, topological analyses were performed to highlight critical miRNAs and their associated target genes. The potential roles of these key miRNAs in L. major infection were further explored and discussed.
3. Results
3.1. Protein Homolog Among L. major, Insects, and Mammals
After performing protein sequence alignments, duplicate homologs between L. major and eight mammalian species were removed. Among mammals, human proteins shared the highest homology with L. major, with 3785 homologous proteins identified. The lowest number of homologs was observed in O. cuniculus (rabbit), with 3465 proteins. Overall, the proportion of homologous proteins between L. major and the mammalian proteomes ranged from 40% to 45% (Figure 2A). Understanding these differences between pathogen and host may inform the development of drugs that specifically target the protozoan while minimizing host toxicity.
Comparisons with ten insect species revealed that most homologous protein percentages were approximately 40% (Figure 2B), e.g., D. melanogaster (39.94%) and A. gambiae (40.27%). Notably, P. papatasi, the primary vector of L. major, exhibited the lowest proportion of homologs with this protozoan. These results indicate that the majority of L. major proteins are significantly distinct from those of mammals and insects.
A two-sample t-test was conducted using Origin software (version 10.1) to evaluate whether mean homologous protein counts differed significantly between mammals and insects. The null hypothesis assumed no difference, while the alternative hypothesis assumed a significant difference. The results indicated p-values ≤ 0.01 (Figure 2C), suggesting a statistically significant difference in homologous protein quantities between mammalian and insect groups. For the mammalian group, the standard error of the mean (SEM) was 36.68 and the 95% confidence interval (CI) was [3627, 3800.5]; for insects, SEM was 38.7 and CI was [3287.46, 3462.54]. Although overall homology proportions are relatively low, further functional analyses may elucidate the roles of these proteins in protozoan survival, host adaptation, and disease progression, providing potential targets for therapeutic development.
Based on sequence alignments, the occurrence frequencies of 4149 homologous proteins across all 18 species (8 mammals and 10 insects) were calculated and visualized (Figure 2D). The results revealed notable differences in protein distribution between species: 3813 proteins were present in at least six species, whereas 2459 proteins were conserved across all 18 species. This distribution provides a foundation for investigating potential host–protozoan interactions and may guide future studies into the molecular mechanisms of L. major infection.
3.2. Protein Distribution Among L. major, P. papatasi, and Humans
Following BLAST sequence alignments, we conducted a detailed analysis of homolog distribution in P. papatasi and humans, representing the insect vector and mammalian host, respectively. Among insect homologs, 13 proteins were absent in all mammals, and 34 proteins were absent specifically in humans (Figure 3A). For P. papatasi, 46 proteins were not found in mammals, while 93 proteins were absent in humans. Conversely, among mammalian homologs, 100 proteins were absent in insects and 497 proteins were absent in P. papatasi (Figure 3B). These findings suggest that L. major proteins exhibit differential similarities across hosts, potentially reflecting variations in host immune responses to protozoan infection.
A Venn diagram illustrating protein homolog distribution across L. major, Homo sapiens, and P. papatasi revealed that 2963 proteins were shared among all three species, while 4659 proteins were unique to L. major (Figure 3C). The number of homologous proteins shared with humans was notably higher than with P. papatasi, which may indicate greater adaptability of L. major to mammalian hosts. These results provide insights into future studies on the complex mechanisms underlying L. major virulence in humans.
3.3. Function Enrichment Analysis of Homologous Proteins
Sequence alignments between the proteomes of eight mammals and L. major were performed, and the intersection set of proteins present in all mammals was identified using the bi-directional best hit strategy. Gene Ontology (GO) enrichment analysis of these conserved proteins was conducted using DAVID, with a significance threshold of p ≤ 0.05. GO terms were categorized into three classes—biological process, cellular component, and molecular function—and visualized using pie charts (Figure 4).
In the biological process category, “mRNA splicing via spliceosome (GO:0000398)” accounted for the largest proportion (17.6%). This process is a critical step in regulating gene expression via processing precursor mRNAs into mature transcripts, thereby influencing protein synthesis and function [24]. Within the cellular component category, “cytosol (GO:0005829)” represented the highest proportion (24.2%), suggesting that many L. major proteins function within host macrophage-like cells during infection [25]. In the molecular function category, “protein binding (GO:0005515)” dominated (78%), highlighting the central role of these proteins in mediating protein–protein interactions and potentially participating in key biological processes such as signal transduction and metabolic regulation [26].
These GO enrichment results provide a comprehensive overview of the functional distribution of L. major and mammalian homologous proteins. They offer insights into the molecular mechanisms underlying protozoan virulence and host interactions and help to identify functional differences that may contribute to disease development.
3.4. Annotation of Hypothetical Proteins
A total of 2346 hypothetical proteins of L. major were retrieved from the NCBI database in February 2025. Novel annotations were assigned based on sequence homology to the best-matching proteins identified through BLAST alignment. Using this approach, 94 hypothetical proteins were successfully annotated (Table S1), with the 15 proteins exhibiting the lowest E-values highlighted in Table 1.
For example, protein XP_001686672.1, previously annotated as “conserved hypothetical protein,” was re-annotated as “Intraflagellar Transport Protein 122” with an extremely low alignment E-value of 2.27 × 10^−149^. In UniProt, this protein remains listed as hypothetical, whereas TriTrypDB annotates it as intraflagellar transport protein 122. Similarly, protein XP_888594.1 was annotated as “Tetratricopeptide Repeat Domain 21B” with an E-value of 1.57 × 10^−117^.
These results demonstrate that homology-based annotation can provide reliable functional insights into previously uncharacterized proteins. Further experimental validation (“wet lab” studies) is warranted to confirm these annotations. Overall, this analysis enhances our understanding of potential mechanisms underlying host adaptation and infection by L. major.
3.5. Infection-Related Protein Analysis
Previous studies have demonstrated that L. major relies on key proteins, including tyrosine phosphatases, cytokines, and kinases, to infect mammalian hosts [16,17,18]. Based on our protein homology analysis, proteins annotated with keywords such as “tyrosine phosphatase,” “cytokine,” or “kinase” were extracted, and detailed results are presented in Table 2. No novel cytokines were identified in L. major through homology searches. However, two infection-related protein groups—kinases and tyrosine phosphatases—were newly annotated. In total, 24 protein kinases and three tyrosine phosphatases were identified within the L. major proteome. Given their critical roles in signal transduction, these proteins may play essential functions during protozoan infection. For instance, four adenylate kinases (ADK2, ADK7, ADK8, ADK9) were newly identified. Adenylate kinase is a phosphotransferase enzyme that catalyzes the interconversion of adenine nucleotides, suggesting that L. major may depend heavily on cellular energy homeostasis to establish infection [27]. These infection-related proteins represent potential drug targets. Inhibition or functional disruption of these kinases and phosphatases could interfere with protozoan growth and virulence, providing avenues for the development of novel therapeutic interventions.
3.6. Novel miRNA Analysis
The miRNAs are conserved non-coding RNAs that play crucial roles in post-transcriptional regulation. Using an EST-based approach, which has proven effective for identifying novel miRNAs in published genomes, we compared L. major sequences against known miRNAs in the miRBase database and identified a total of 29 miRNAs in the L. major genome. The characteristics of these miRNAs, including length, EST accession, and mismatch values, are summarized in Table 3. The miRNA lengths ranged from 18 to 22 nucleotides, consistent with typical miRNA sizes. Mismatch values are critical for distinguishing miRNAs from other non-coding RNAs. Two miRNAs, lma-miR-9984 and lma-miR-146a, exhibited zero mismatches, indicating complete identity with known miRNAs. Eight miRNAs differed by only a single nucleotide (mismatch = 1) from their known counterparts.
Screening miRNAs using a minimum free energy frequency (FMFE) threshold of ≤0.05 revealed that most predicted MFEI values were significantly negative, supporting the stability of these sequences. Furthermore, the majority of miRNAs exhibited well-formed secondary hairpin structures, providing additional evidence of their reliability and potential functionality.
3.7. Analysis of miRNA Phylogenetic Tree
To investigate the evolutionary relationships among the 29 identified miRNAs, a phylogenetic tree was constructed (Figure 5). In this tree, miRNAs clustered within the same branch are inferred to share a common ancestor, while branch lengths and node positions reflect the extent of evolutionary changes. The phylogenetic tree can be clearly divided into two main branches: the first branch ranges from lma-miR-4079-3p to lma-miR-4272, and the second branch ranges from lma-miR-2227 to lma-miR-1237. Highly conserved miRNAs often cluster within sub-branches; for instance, lma-miR-3578 to lma-miR-9984 form a sub-branch, suggesting they may retain similar functions across species.
Support values indicated on the tree represent the frequency with which a node or branch appears across multiple reconstructions. Notably, two miRNAs, lma-miR-7008-3p and lma-miR-6886-3p, exhibited the highest support values of 67, indicating that these nodes are highly reliable in representing L. major miRNA genome evolution.
3.8. Distribution of Identified miRNAs in Mammals
The identified L. major miRNAs were compared with homologous sequences in the miRBase database to examine their distribution across mammalian and non-mammalian species (Table 4). Seven miRNAs were found to be present only in non-mammalian species; for example, miR-10117-3p was detected exclusively in nematode H. polygyrus, suggesting a potential functional similarity in host-protozoan interactions between L. major and H. polygyrus. Conversely, miR-3960 and miR-150-3p were present in both H. sapiens and M. musculus, indicating that these miRNAs may perform conserved functions in mammals. Additionally, miR-146a was detected in four mammalian species (B. taurus, P. troglodytes, E. caballus, and C. jacchus), highlighting its potential as a highly conserved and functionally important miRNA. These findings provide a foundation for further investigation into the roles of these miRNAs in host-protozoan interactions and the pathogenic mechanisms of L. major.
3.9. Analysis of miRNA-Gene Interaction Network
To further investigate the regulatory roles of L. major miRNAs, we identified miRNA-targeted genes and constructed a miRNA-gene interaction network using Cytoscape (Figure 6). In this network, six key miRNAs were found to interact with 23 proteins. Notably, lma-miR-142-5p exhibited the highest number of target genes, suggesting its role as a central regulator potentially involved in multiple biological processes. Several proteins in the network represent critical nodes. For example, superoxide dismutase 2 (SOD2) is essential for maintaining mitochondrial integrity and function, as mitochondria are the primary sites of reactive oxygen species (ROS) production during cellular respiration. SOD2 thus protects cells from oxidative damage, regulates apoptosis, and contributes to overall cellular homeostasis [28]. HSPA8, a member of the heat shock protein 70 (Hsp70) family, functions as a molecular chaperone involved in protein folding, stress response, and signal transduction, playing a crucial role in maintaining protein homeostasis and cell survival under various conditions [29]. These findings emphasize the importance of studying the 23 proteins within the network to elucidate their potential roles in the infection mechanisms of L. major.
4. Discussion
According to World Health Organization (WHO), leishmaniasis affects populations in endemic areas, with an estimated 700,000–1,000,000 new cases and approximately 20,000–30,000 deaths per year worldwide [30]. The current L. major genome comprises 8,537 proteins, of which 2319 have unknown functions. To explore the potential roles of these proteins, we performed sequence alignments between L. major and the proteomes of eight mammalian species. H. sapiens exhibited the highest level of homology, with 3,785 proteins shared. Overall, homologous proteins accounted for 40–45% of the L. major proteome across these mammals. A two-sample t-test revealed a highly significant difference (p ≤ 0.01) in the number of homologous proteins between mammals and insects. Based on these results, we successfully annotated 116 hypothetical proteins, which laid the foundation for subsequent functional analyses. Functional enrichment analysis revealed that “mRNA splicing via spliceosome (GO:0000398)” represented 17.6% of the biological process category, whereas “protein binding (GO:0005515)” accounted for 78% in the molecular function category, indicating a key role in mediating protein–protein interactions.
By aligning against the NCBI non-redundant database, we further annotated 94 hypothetical proteins. Among these, 27 infection-related proteins were identified, including 24 protein kinases and three tyrosine phosphatases. Eukaryotic protein kinases have been reported as promising targets for antileishmanial drug development due to their crucial roles in signal transduction via phosphorylation. Inhibiting these kinases is expected to disrupt essential processes such as cell-cycle progression, differentiation, and virulence, ultimately affecting Leishmania viability and parasitic survival.
While protein kinases and miRNAs emerged as promising therapeutic candidates, the issue of target specificity warrants careful consideration. Because many eukaryotic kinases are evolutionarily conserved [31], cross-reactivity with host kinases remains a potential risk. However, several of the kinases identified here show Leishmania-specific sequence motifs and domain architectures distinct from their mammalian counterparts. For instance, multiple kinase domains display insertions, deletions, or divergent activation-loop residues unique to trypanosomatids, suggesting that selective inhibition may be achievable. Future structural modeling and comparative docking analyses could help pinpoint parasite-specific pockets that differ from those in host homologs. Similarly, transcriptomic and proteomic profiling during infection can further verify parasite-specific expression, minimizing off-target effects on host cells. Collectively, integrating sequence, structural, and expression data will be critical to prioritize Leishmania-specific targets and enhance the translational relevance of these findings.
In addition, 29 miRNAs were identified in the L. major genome by comparison with known sequences in miRBase. Phylogenetic analysis divided these miRNAs into two distinct branches: the first from lma-miR-4079-3p to lma-miR-4272, and the second from lma-miR-2227 to lma-miR-1237. Homology analysis revealed that seven miRNAs (lma-miR-10117-3p, lma-miR-2227, lma-miR-2g-5p, lma-miR-10849, lma-miR-4171-5p, lma-miR-4079-3p, and lma-miR-4057-3p) were absent in all mammals, suggesting their potential as protozoa-specific regulatory molecules and targets for drug development.
The miRNA-gene interaction network further highlighted key regulatory genes, including MAPK1 and GSK3B. MAPK1 (Mitogen-Activated Protein Kinase 1) plays a central role in signal transduction, transmitting extracellular stimuli from the cell surface to the nucleus [32,33]. GSK3B (Glycogen Synthase Kinase 3 Beta) is a serine/threonine kinase involved in diverse cellular processes, including autophagy and glycogen metabolism. It phosphorylates and inhibits glycogen synthase, thereby regulating glucose storage [34,35]. These findings underscore the presence of critical genes and pathways associated with L. major infection and provide a foundation for future studies on host–parasite interactions and therapeutic interventions.
Importantly, our integrated analysis of protein-coding genes and miRNAs provides a comprehensive view of the molecular machinery underlying L. major virulence. By combining functional annotation, enrichment analysis, and miRNA-gene network construction, we identified protozoan-specific molecules and infection-related pathways that may serve as novel drug targets or biomarkers. This study not only enhances our understanding of the molecular basis of L. major infection but also establishes a framework for future experimental validation and therapeutic development.
Our study offers valuable insights into the complex interplay among L. major, its insect vector, and mammalian hosts. The identification of novel miRNAs and infection-related proteins opens new avenues for antileishmanial drug discovery, while the functional and evolutionary analyses provide a foundation for understanding protozoan adaptation and host specificity. Future research should focus on experimental validation of these targets, the mechanistic roles of identified miRNAs, and the development of targeted interventions to mitigate the global burden of cutaneous leishmaniasis.
In addition to parasite-derived molecular targets, vector–parasite interactions represent a critical but often underexplored aspect of leishmaniasis transmission. P. papatasi, the principal vector of L. major, exhibits species-specific molecular determinants, such as midgut receptors and salivary proteins, that facilitate parasite attachment and survival [36]. Comparative analyses of vector transcriptomes and proteomes have revealed unique signatures that could serve as biomarkers for potential targets to interrupt transmission. Integrating these vector-specific findings into therapeutic and diagnostic frameworks, such as designing inhibitors that disrupt parasite-vector binding, may enhance the translational potential of Leishmania research. Future work should thus aim to combine parasite and vector omics data to better understand and control leishmaniasis at the molecular interface between parasite and vector.
To experimentally validate our computational predictions, several complementary strategies can be employed. First, the parasite-specific miRNAs identified in this study could be functionally characterized using gain- and loss-of-function assays, followed by quantitative assessment of parasite growth and infectivity. The downstream target genes predicted from the miRNA-gene interaction network could be verified through dual-luciferase reporter assays and RT-PCR analyses to confirm regulatory relationships [37]. Second, kinase inhibition studies using specific small-molecule inhibitors could help determine the functional importance of the identified infection-related kinases in protozoan viability and virulence. Third, macrophage infection models could be applied to evaluate how modulation of these miRNAs or kinase genes affects host–parasite interactions, cytokine responses, and intracellular parasite survival. Together, these approaches will provide direct evidence supporting the bioinformatic predictions and strengthen the mechanistic understanding of L. major virulence.
5. Conclusions
We identified novel protein-coding genes and miRNAs in Leishmania major through comparative genome analysis with its insect vector and mammalian hosts. Several infection-related proteins and miRNAs were highlighted as potential biomarkers and drug targets. These findings advance our understanding of L. major virulence and provide a foundation for future antileishmanial therapy development.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mcghee S. Angus N. Finnegan A. Lewis-Pierre L. Ortega J. Assessment and treatment of cutaneous leishmaniasis in the emergency department Emerg. Nurse 202328232910.7748/en.2020.e 199332017482 · doi ↗ · pubmed ↗
- 2Piyasiri S.B. Dewasurendra R. Samaranayake N. Karunaweera N. Diagnostic Tools for Cutaneous Leishmaniasis Caused by Leishmania donovani: A Narrative Review Diagnostics 202313298910.3390/diagnostics 1318298937761356 PMC 10529649 · doi ↗ · pubmed ↗
- 3Kaye P.M. Matlashewski G. Mohan S. Le Rutte E. Mondal D. Khamesipour A. Malvolti S. Vaccine value profile for leishmaniasis Vaccine 202341 S 153S 17510.1016/j.vaccine.2023.01.05737951693 · doi ↗ · pubmed ↗
- 4Ivens A.C. Peacock C.S. Worthey E.A. Murphy L. Aggarwal G. Berriman M. Sisk E. Rajandream M.A. Adlem E. Aert R. The genome of the kinetoplastid parasite, Leishmania major Science 200530943644210.1126/science.111268016020728 PMC 1470643 · doi ↗ · pubmed ↗
- 5Camacho E. González-De La Fuente S. Solana J.C. Rastrojo A. Carrasco-Ramiro F. Requena J.M. Aguado B. Gene annotation and transcriptome delineation on a de novo genome assembly for the reference Leishmania major Friedlin strain Genes 202112135910.3390/genes 1209135934573340 PMC 8468144 · doi ↗ · pubmed ↗
- 6Yang Z. Zeng X. Tsui K.W.S. Investigating function roles of hypothetical proteins encoded by the Mycobacterium tuberculosis H 37Rv genome BMC Genom.20192039410.1186/s 12864-019-5746-6PMC 652828931113361 · doi ↗ · pubmed ↗
- 7Sundararaj R. Mathimaran A. Prabhu D. Ramachandran B. Jeyaraman J. Muthupandian S. Asmelash T. In silico approaches for the identification of potential allergens among hypothetical proteins from Alternaria alternata and its functional annotation Sci. Rep.202414669610.1038/s 41598-024-55463-138509156 PMC 10954717 · doi ↗ · pubmed ↗
- 8Nimsarkar P. Ingale P. Singh S. Systems studies uncover mi R-146a as a target in leishmania major infection model ACS Omega 20205125161252610.1021/acsomega.0c 0150232548436 PMC 7271362 · doi ↗ · pubmed ↗