Enantioselective Preparation of (N,O)-, (N,N)- and (N,S)-Spiroketal Moieties
Mata Sow, Edouard Fauran, Laurent Commeiras

TL;DR
This review discusses methods for creating specific spiroketal structures with high enantioselectivity, important in natural products and drugs.
Contribution
The paper provides a comprehensive overview of enantioselective strategies for synthesizing (N,O)-, (N,N)-, and (N,S)-spiroketal moieties.
Findings
Enantioselective strategies rely on metallo-, organo-, or Lewis acid-catalyzed reactions.
These methods are effective for constructing spiroketal skeletons found in natural products and pharmaceuticals.
Abstract
This review highlights enantioselective strategies for preparing (N,O)- (N,N)- and (N,S)-spiroketal skeleton, which are key structural units found in many natural products and pharmacologically active substances. The strategies are mainly based on metallo-, organo- or Lewis acid-catalyzed cycloaddition or annulation reactions.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
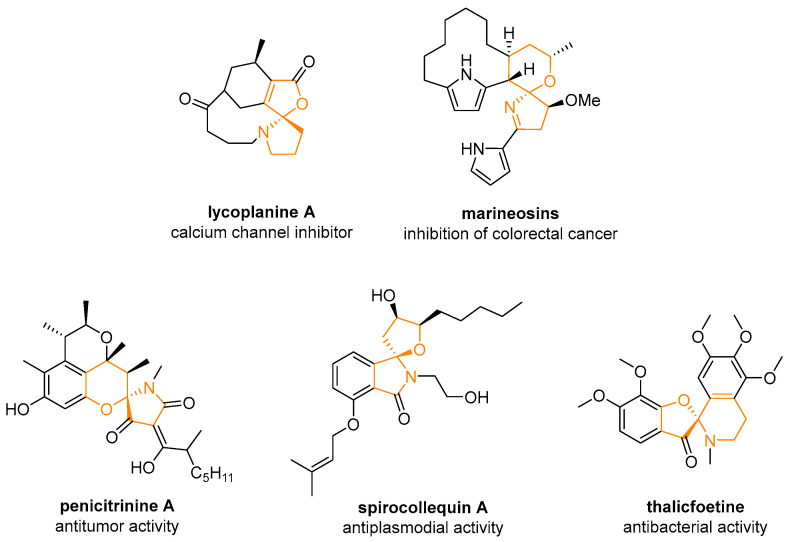 Figure 1
Figure 1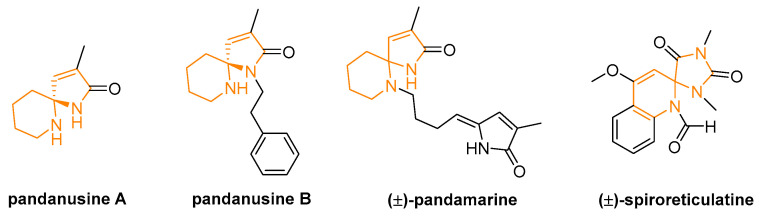 Figure 2
Figure 2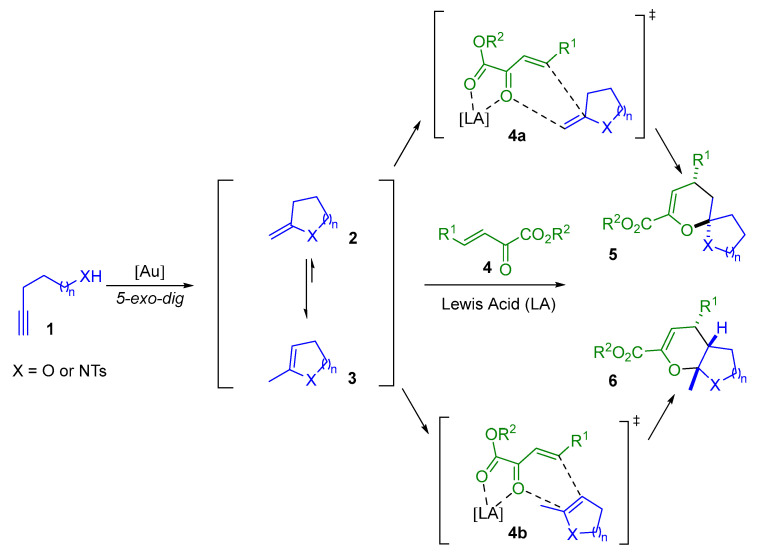 Figure 3
Figure 3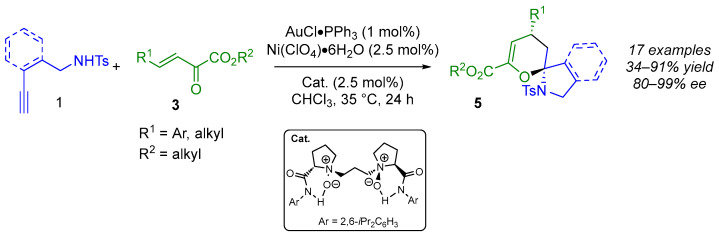 Figure 4
Figure 4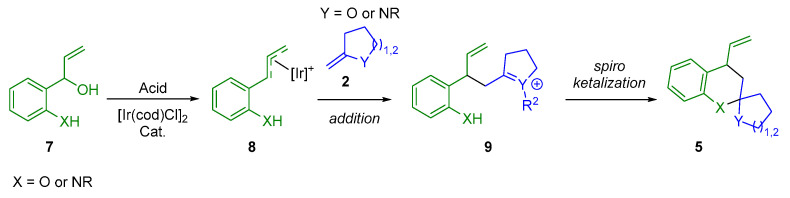 Figure 5
Figure 5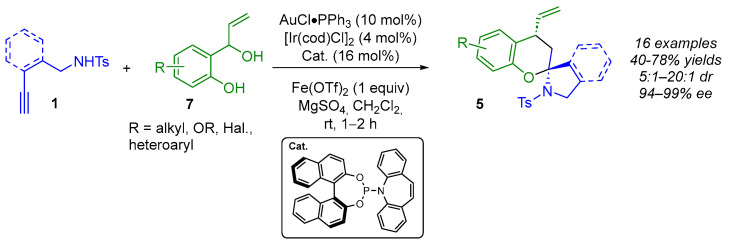 Figure 6
Figure 6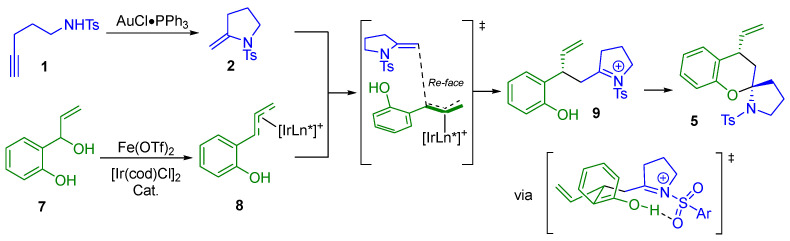 Figure 7
Figure 7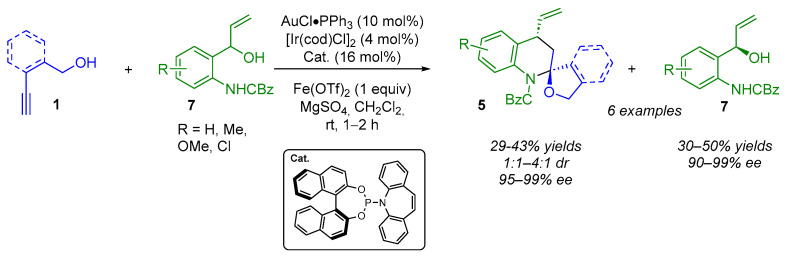 Figure 8
Figure 8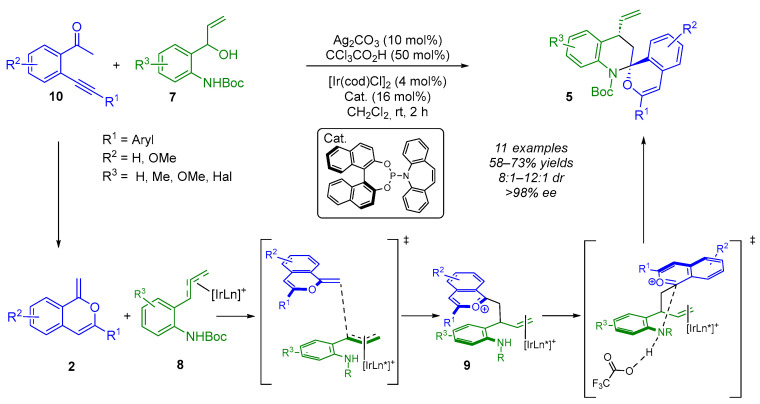 Figure 9
Figure 9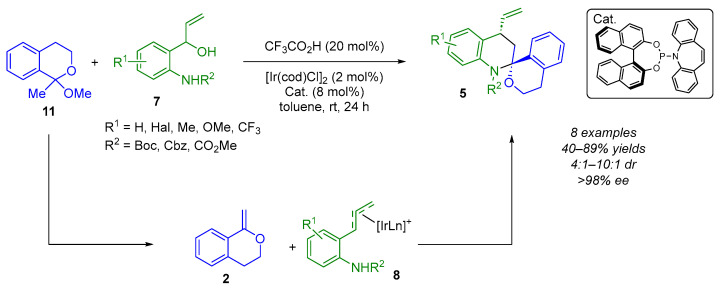 Figure 10
Figure 10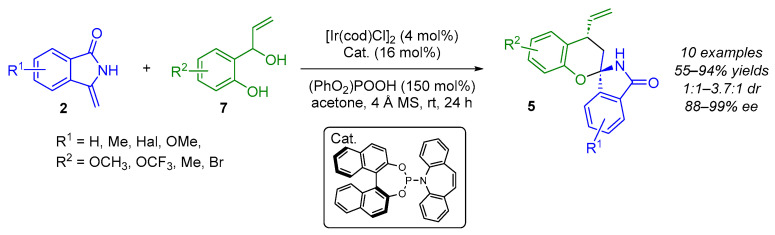 Figure 11
Figure 11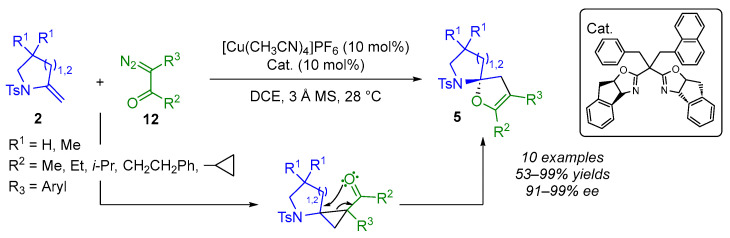 Figure 12
Figure 12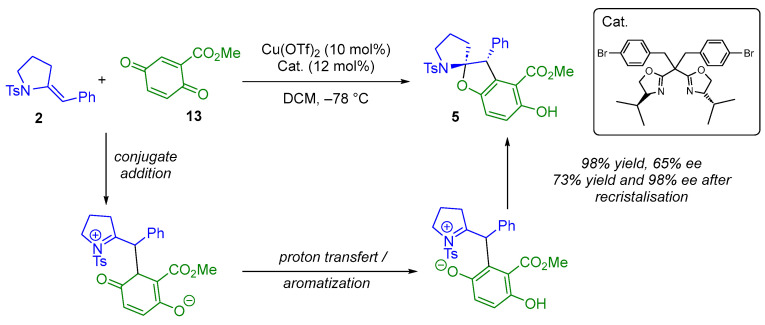 Figure 13
Figure 13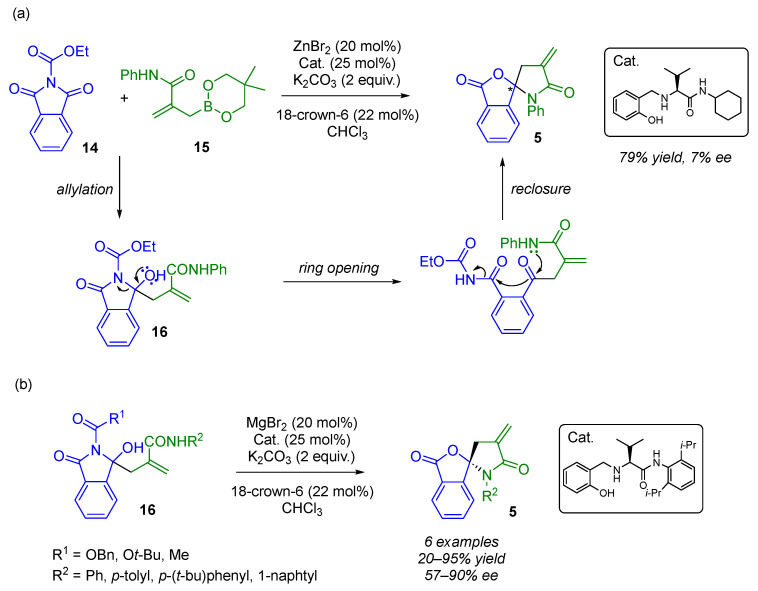 Figure 14
Figure 14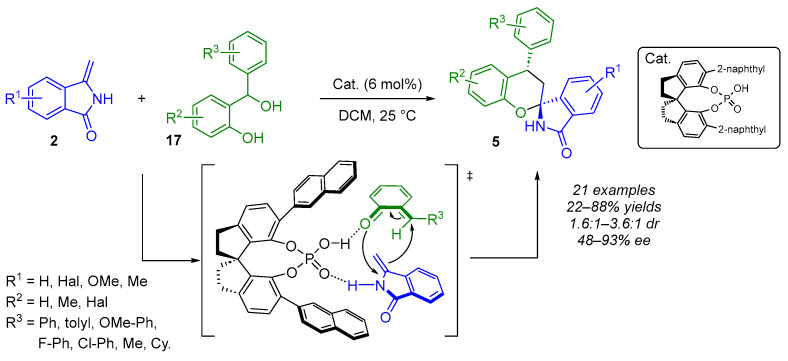 Figure 15
Figure 15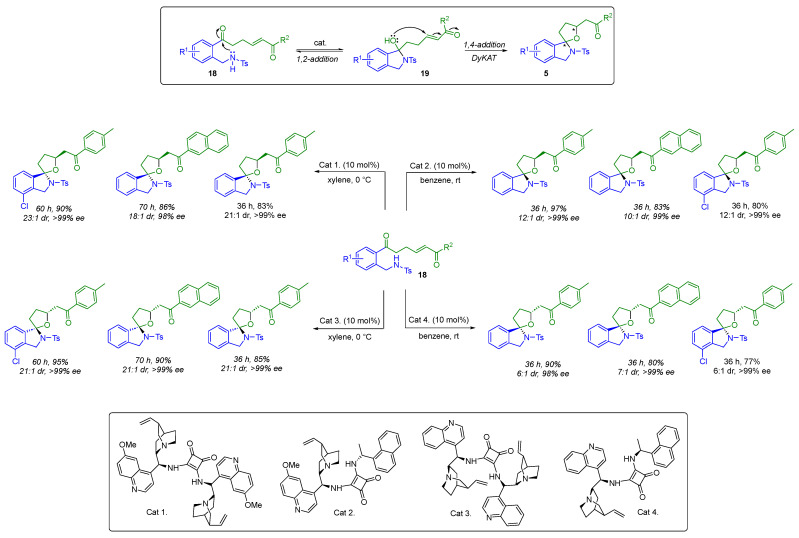 Figure 16
Figure 16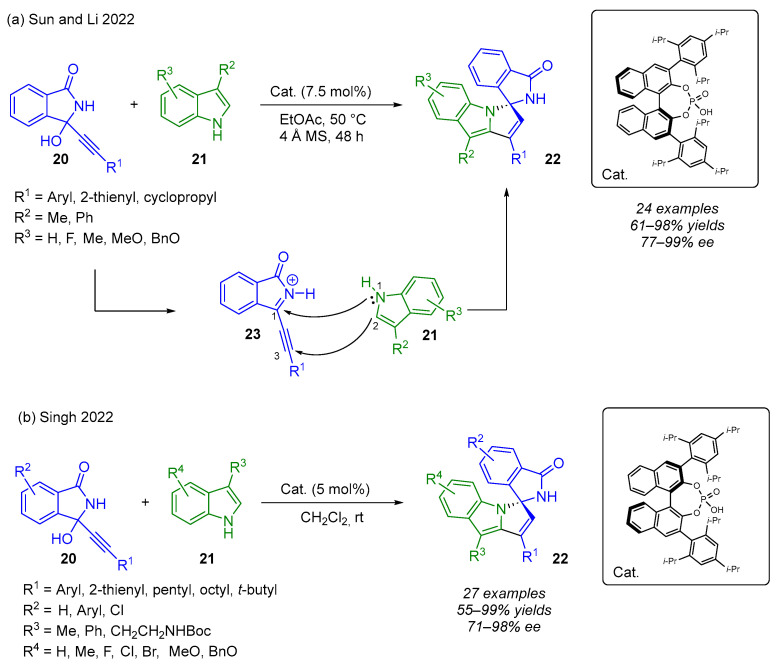 Figure 17
Figure 17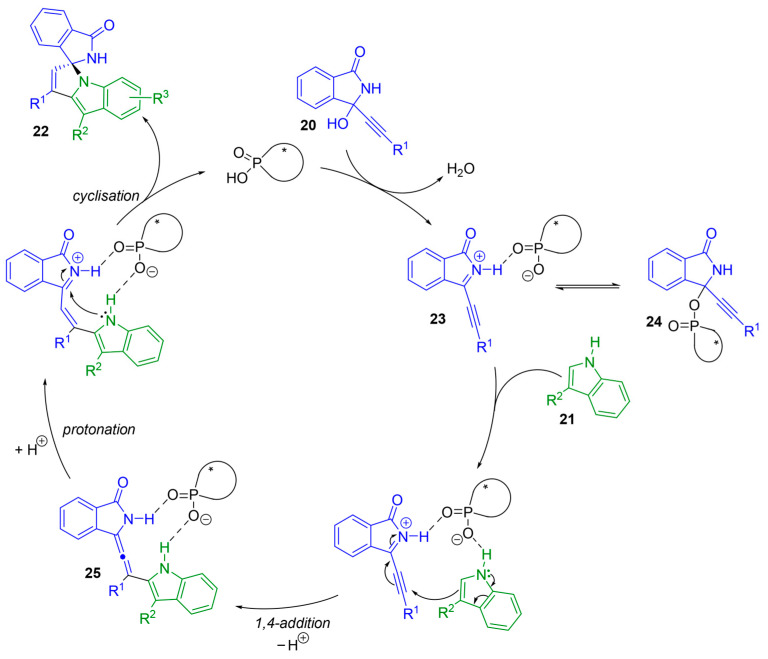 Figure 18
Figure 18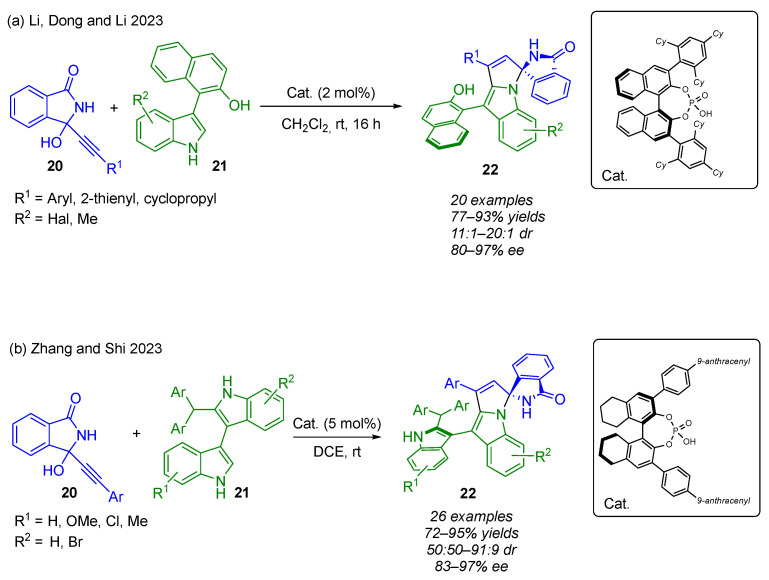 Figure 19
Figure 19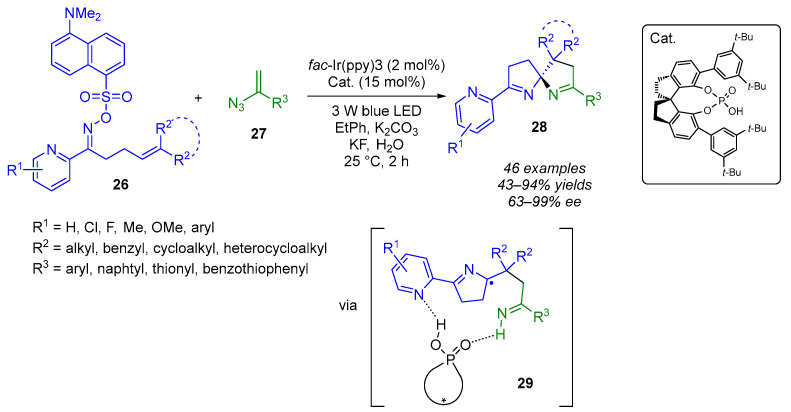 Figure 20
Figure 20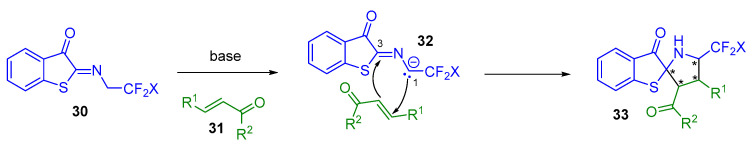 Figure 21
Figure 21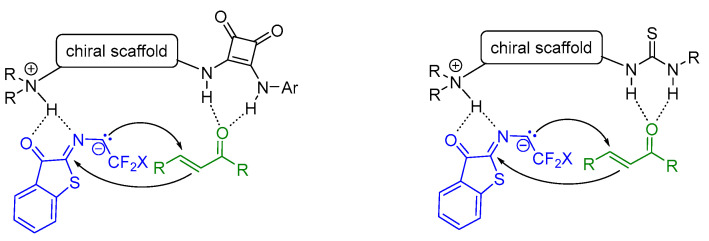 Figure 22
Figure 22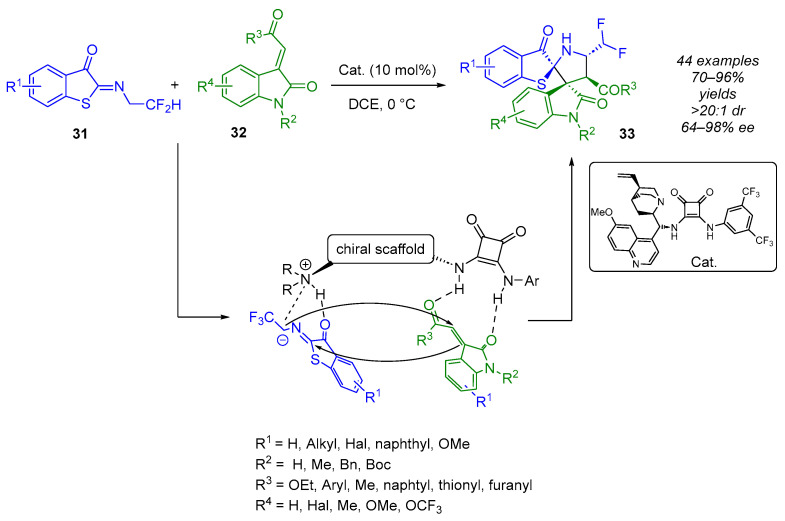 Figure 23
Figure 23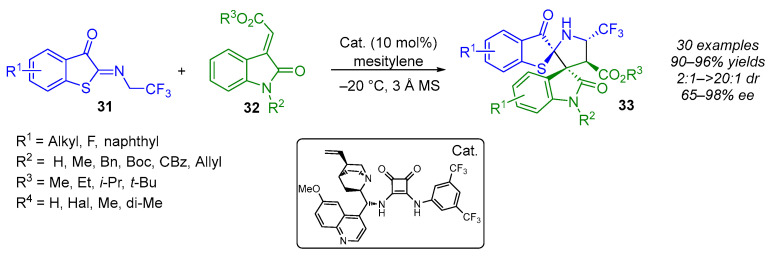 Figure 24
Figure 24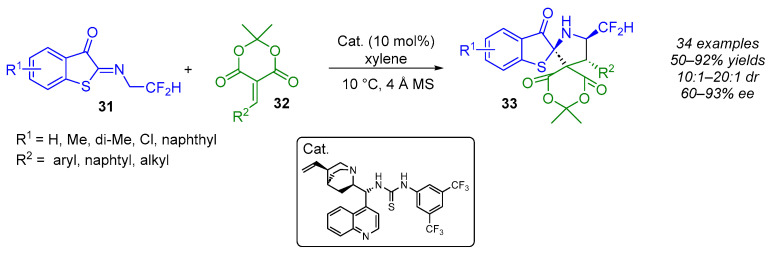 Figure 25
Figure 25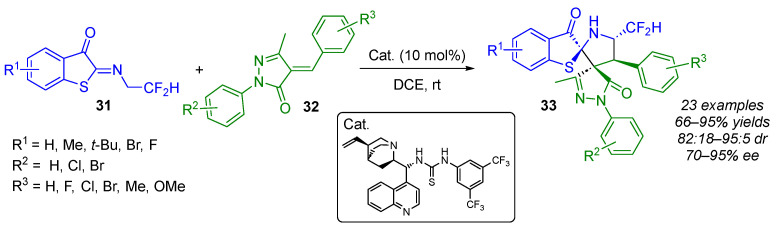 Figure 26
Figure 26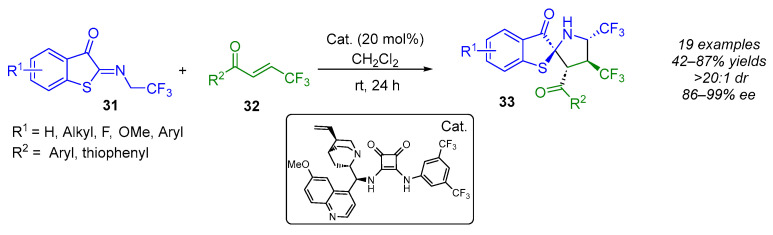 Figure 27
Figure 27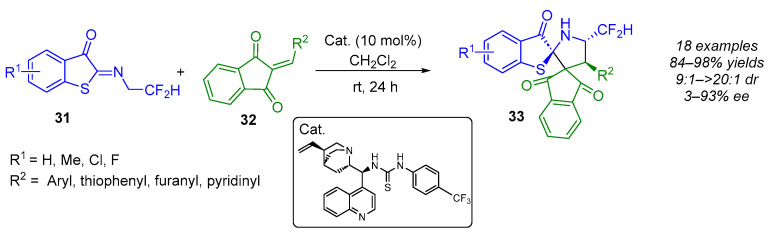 Figure 28
Figure 28Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCarbohydrate Chemistry and Synthesis · Lipid metabolism and biosynthesis
1. Introduction
According to the International Union of Pure and Applied Chemistry (IUPAC), spirocyclic compounds are defined as “compounds having one atom (usually a quaternary carbon) as the only common member of two rings” [1]. These molecules have attracted considerable interest due to the unique properties imparted by the spirocyclic quaternary carbon stereocenter, which confers conformational rigidity to the structure [2,3,4,5,6,7,8]. Since nitrogen-containing heterocyclic scaffolds represent a crucial class of compounds, it is not surprising that a wide variety of aza-spiroheterocyclic molecules are found in biologically active compounds and natural products. Among them, (N,O)-spiroketal derivatives are no exception (Figure 1). Compared to racemic strategies for the construction of such spiroaminals [9], the enantioselective construction of (N,O)-spirocenters is challenging due to its instability and propensity for racemization, even in mildly acidic conditions. In this review, we would like to describe progress in the field of enantioselective synthesis of such derivatives, as well as less studied (N,N)- and (N,S)-spiroketals.
2. (N,O)-Spiroketals
(N,O)-Spiroketal scaffolds represent a distinct class of spiroheterocyclic frameworks found in both natural and synthetic bioactive molecules. Their uniquely rigid three-dimensional architecture, along with their capacity to influence the physicochemical and pharmacokinetic properties of drug candidates, has driven increasing interest among synthetic and medicinal chemists. This has motivated the development of enantioselective strategies, including metallo-, Lewis acid- and organocatalyzed activation, for constructing these challenging spiroheterocyclic moieties.
2.1. Metallocatalyzed [4 + 2] Cycloaddition Reaction
Alkynyl alcohols and amides 1 are versatile precursors that can undergo 5-exo-dig cyclization in the presence of a gold(I) catalyst to generate enol-ether or enamine-ether intermediates 2. These electron-rich alkene intermediates can be engaged in situ in an inverse electron-demand hetero-Diels–Alder reaction (IED hetero-DA reaction) with Lewis acid-activated α,β-unsaturated keto esters 4, leading to the formation of (O,O)- or (N,O)-spiroketals 5. A key challenge in this reaction is the control of the isomerization of the intermediate 2 to form the more thermodynamically stable species 3 (Scheme 1). This process can lead to the formation of fused (O,O)- or (N,O)-ketals 6 instead of the spirocyclic products 5 [10,11,12,13].
In 2016, Liu and Feng demonstrated that introducing a bulkier chiral ligand to the Lewis acid favored the reactivity of the less sterically hindered enamine-ether 2 over 3 in the IED hetero-DA reaction. This shifted the isomerization equilibrium towards 2, thereby enhancing the formation of the desired spirocyclic products. Therefore, using an enantioselective relay catalytic system based on the combination of achiral π-acid gold(I) catalysis and chiral Lewis acid N,N’-dioxyde nickel(II) catalysis (Scheme 2), the authors obtained the desired spiroaminal 5 in good yields (34–91%), with high diastereoselectivity and excellent enantioselectivity (96–99%) [14]. It is worth noting that alkynyl amides were found to be more reactive than alkynyl alcohols, and that the spiroaminal products were obtained with higher yields and greater diastereo- and enantioselectivities. The lowest enantioselectivity (80%) was observed with phenyl-substituted alkynyl amide 1.
It is worth noting that Kang reported a gold(I)/rhodium(III) bimetallic relay catalysis to furnish the spiroaminal using a similar strategy. Unlike spiroketals, which were obtained with excellent enantioselectivity, this approach was less effective for spiroaminal moieties [15].
2.2. Metallocatalyzed (4 + 2) Annulation Reaction
In the previous example (Scheme 2), an IED hetero-DA reaction involving vinyl enamides 2 and α,β-unsaturated keto esters 3 was used to form spiroaminal moieties 5. Another method of accessing these motifs involves a cascade reaction comprising an addition reaction followed by a spiroacetalization reaction (Scheme 3) [16,17,18,19]. This methodology involves a reaction between a nucleophile 2 (an exocyclic vinyl enol ether or an exocyclic vinyl enamine) and an amphiphilic compound 8 that has both an electrophilic and a nucleophilic site. These amphiphilic species 8, which are chiral π-allyl-Ir intermediates, can be obtained via iridium-mediated dehydration of 2-(1-hydroxyallyl)phenols or 2-(1-hydroxylallyl)anilines 7 in the presence of an acid promoter.
In 2022, Deng and Yang investigated the use of amphiphilic reagents alongside alkynyl amides to create (N,O)-spiroketals 5 (Scheme 4) [16]. In the presence of in situ-generated gold(I)-catalyzed vinyl enamides 2 from 1, the desired spiroaminal products 5 were obtained via an enantioselective addition/spiroacetalization process. Under optimized conditions, 5 were obtained with good yields (40–78%) and high diastereo- and enantioselectivities (5:1–20:1 dr, 94–99% ee). The lowest diastereoselectivity (5:1–10:1) was observed when the aromatic ring of 7 was substituted in position 3.
From a mechanistic point of view (Scheme 5), the first step is the gold(I)-catalyzed 5-exo-dig intramolecular hydroamination, which produces the corresponding exocyclic enamide 2. Simultaneously, in the presence of Fe(OTf)2, the chiral Ir(I) complex would produce chiral π-allyl-Ir intermediates 8, which would react with the exocyclic enamine to give the allylation intermediate 9. Due to the shielded Si-face of 7, enamide 2 would approach 7 from the Re-face. Finally, the spirocyclization step would occur via a chair-like Zimmerman–Traxler transition state, giving the desired (N,O)-spiroketal 5.
Interestingly, when using 2-(1-hydroxylallyl)anilines 7 as pre-amphiphilic species and alkynyl alcohols 1 (Scheme 6), the authors observed a kinetic resolution of 7 leading to both spiroaminal moieties 5 (with average diastereoselectivities of 2:1–4:1 and excellent enantioselectivities of 95–99% ee) and the enantiomerically enriched (90–99% ee) residual alcohols 7.
The same group extended this methodology for use with electron-rich methyleneisochromenes 2 as exocyclic enol ethers (Scheme 7). These are formed in situ from ortho-alkynylacetophenones 10 via a 6-endo-dig cycloisomerization reaction followed by a proton transfer step [17]. In the presence of the iridium-π-allyl-amino-dipole intermediate 8, bisbenzannulated spiroaminals 5 are obtained in an extremely stereoselective manner (8:1–12:1 dr, >98% ee). Unlike the previous example, this challenging Ir/Ag/acid ternary catalysis does not lead to kinetic resolution of the starting 2-(1-hydroxylallyl)anilines 7. Moreover, mechanistic investigations and theoretical calculations revealed that the enantioselectivity (allylation step) was controlled by the chiral iridium catalyst, while the diastereoselectivity was attributed to thermodynamically promoted spiroamination, catalyzed by a Brønsted acid.
Despite the flexibility and modularity of the precedented reported methodologies, the use of a bi- or trimetallic catalytic system increases the complexity of the process. Indeed, all of the above examples use exocyclic enol ethers or exocyclic enamines 2 formed in situ via metal catalysis. However, in the following example, Yang and Zheng used isochromane ketals 11, which produce exocyclic enol ethers 2 in an acidic environment, eliminating the need for a metal-catalyzed cycloisomerization reaction (Scheme 8) [18]. In the presence of [Ir(cod)Cl]2, Carreira’s P-olefin ligand and CF_3_CO_2_H, the cascade reaction proceeded smoothly, providing the (N,O)-spiroketal products 5 with moderate diastereoselectivities (4:1–10:1 dr) and excellent enantioselectivities (>98% ee) in 40–89% yields.
In 2024, Liu and Li developed a similar strategy involving 3-methylene isoindolones 2 (Scheme 9) [19]. These compounds are interesting because they are easily accessible and stable enamide moieties. After determining the optimal conditions, the authors demonstrated that this strategy could efficiently produce the desired spiroaminal moieties 5, regardless of the nature of the substituents (electron-withdrawing, electron-donating or bulky groups) present on the isoindolinones 2 and/or pre-amphiphilic 2-(1-hydroxyallyl)phenols 7.
2.3. Metallocatalyzed (3 + 2) Annulation Reaction
Another proven strategy for accessing enantiomerically enriched (N,O)-spiroketal moieties is based on a process consisting of a cyclopropanation reaction followed by a rearrangement (CP-RA). Tang and Wang developed a copper-catalyzed enantioselective CP-RA reaction involving exocyclic enamides 2 and α-aryl-α-diazoketones 12, achieving high levels of selectivity (Scheme 10) [20]. Indeed, both α-diazoketones 12, bearing various carbonyl substituents (such as acetyl, propionyl and isopropyl groups), and five- or six-membered exocyclic enamides 2 have been shown to be suitable substrates. The corresponding products 5 were obtained in good yields (53–99%) and with high enantiomeric excesses (91–96% ee).
1,4-Benzoquinone derivatives are readily available starting materials that have been shown to undergo a conjugate addition reaction with nucleophiles. Following proton transfer and aromatization steps, the resulting phenolate anions formed in situ can engage in a cyclization reaction to produce oxygen-containing five-membered rings. In this context, the same group developed a copper-catalyzed series of annulation reactions using 1,4-benzoquinone derivatives (Scheme 11) [21,22,23]. More specifically, the authors studied the racemic reaction of five- or six-membered exocyclic enamides and sulfonylenamides 2 with 1,4-benzoquinone derivatives 13 in order to construct (N,O)-spiroketal moieties 5 via (3 + 2) annulation reactions. The authors validated their methodology by reporting an enantioselective example. Using a chiral bisoxazoline ligand, they obtained the desired product with a yield of 98% and an enantiomeric excess of 65% (98% after recrystallization) [23].
2.4. Lewis Acid Catalysis
Yoda described the synthesis of new types of biologically active (N,O)-spirocyclic compounds. These were formed via a Lewis acid-catalyzed reaction cascade involving the nucleophilic addition of amide-functionalized allylboronates 15 to N-carbonylimides 14, followed by the opening reaction and the closing reaction of the intermediate hydroxylactam ring 16 [24,25,26]. In 2020, the authors attempted to convert the racemic version into an enantioselective one by adding a chiral ligand, but this proved unsuccessful (Scheme 12a) [27]. Therefore, they simplified the system and began working with γ-hydroxylactam derivatives with a methacrylamide side chain 16 (Scheme 12b). Using a new MgBr_2_-chiral aminophenol-based catalyst, Yoda reported an enantioselective ring-opening and -reclosing cascade smoothly enabling the synthesis of enantiomerically enriched (N,O)-spirocyclic compounds 5 (20–95% yield, 57–90% ee). It was observed that the enantiomeric excess was at its lowest (57% ee) when a bulky substituent, such as tert-butyl group, was attached to the nitrogen atom. This would directly impact the coordination of the substrate with the chiral catalyst.
2.5. Organocatalysis
ortho-Quinone methides (o-QMs) typically act as Michael acceptors and serve as versatile synthetic transient intermediates in catalytic asymmetric conjugate addition and (4 + 2) cycloaddition reactions. One way to obtain o-QMs is to treat ortho-hydroxybenzyl alcohols 17 with a Brønsted acid. From this observation, in 2022, Wang’s group used chiral phosphoric acids as bifunctional catalysts to (1) generate o-QMs from ortho-hydroxybenzyl alcohols 17 and (2) synthesize enantioenriched (N,O)-spiroaminal moieties 5 via a formal (4 + 2) cycloaddition reaction in the presence of 3-methylene isoindolinone 2 (Scheme 13) [28]. Indeed, in the optimal conditions, they obtained the desired spiro chroman-isoindolinones containing (N,O)-spiroketal moieties with moderate diastereoselectivity (1.6:1–3.6:1 dr) and with good enantioselectivities (48–93% ee). During the optimization steps, the authors found that adding the 3-methylene isoindolinone in two batches (60% then 40%) improved the yield of product 5 while maintaining the stereoselectivity. Additionally, the lowest diastereoselectivity (1.6:1 dr) was observed when R^2^ = 4-Br, and the lowest enantioselectivity (48% ee) when R^3^ = m-F-Ph. From a mechanistic point of view, the possibility of a concerted mechanism between 3-methylene isoindolinone and o-QMs could not be ruled out. However, the authors proposed a transition state assembly for the CPA-catalyzed asymmetric formal (4 + 2) cycloaddition, as they suggested that a stepwise Michael addition and subsequent nucleophilic oxygen addition seem more reasonable.
In 2024, Ghorai’s group also exploited a non-covalent interaction to achieve enantioselective synthesis of (N,O)-spiroketal moieties (Scheme 14). Starting with a multifunctional substrate 18, containing a keto-tethered Michael acceptor unit and a protected amine group, they carried out the first stereodivergent synthesis of these spiroheterocycles by using chiral bifunctional organic catalysts [29]. This stereodivergent strategy is based on reversible intramolecular 1,2-addition of the N-protected amine to the carbonyl function, forming the chiral hemiaminal intermediate 19. This is then followed by an oxa-Michael addition via a dynamic kinetic asymmetric transformation (DyKAT). This cascade spirocyclization successfully produced all four highly enantioenriched stereoisomers of spiroisoindolines from the same starting material by modulating the chiral components of squaramide catalysts with a suitable combination of chiral amines. The authors have demonstrated the power and efficiency of this strategy with more than 60 examples. For greater clarity, Scheme 14 only describes examples leading to all stereoisomers. To prove that the second step of spirocyclization is a DyKAT, the authors synthesized a racemic hemiaminal. Treatment with the optimized catalysts provided all the respective stereoisomers with excellent stereocontrol.
3. (N,N)-Spiroketals
Compared with (N,O)-spiroketal units, (N,N)-spiroketal backbones are less prevalent in natural products (see some examples in Figure 2) and have therefore attracted less interest. Nevertheless, several groups have developed enantioselective methods for these spiroaminal structures with the aim of incorporating them into indole and/or isoindoline frameworks. These frameworks represent indispensable structural motifs in numerous pharmaceutical agents that exhibit a wide range of biological activities.
3.1. CPA-Catalyzed (3 + 2) Annulation Reaction
Propargylic alcohols are readily available and versatile synthons in organic synthesis. Their unique bifunctional character makes them valuable intermediates in a range of transformations, including substitutions, conjugate additions and annulations, for the efficient construction of diverse, functionalized molecular architectures [30,31]. Recent advances in stereoselective organocatalysis have enabled the enantioselective transformation of propargylic alcohols, particularly α-functionalized propargylic alcohols (α-FPAs) [32]. More precisely, chiral phosphoric acids (CPAs) and their analogs facilitate the in situ dehydration of α-FPAs to generate electron-deficient intermediates, which undergo highly enantioselective additions. Building on their previous work in CPA-catalyzed reactions of propargylic alcohols, Li and Sun became interested in exploring indolinones as α-auxiliary groups. They achieved an organocatalytic (3 + 2) annulation of 3-alkynyl-3-hydroxyisoindolinones 20 with 1H-indoles 21 affording spiro-isoindolinone-indoline moieties 22 (Scheme 15a) [33]. Strategically, the in situ-generated β,γ-alkynyl-α-ketimine intermediate 23 is comparable to 1,3 di-electrophiles and the 1H-indoles as 1,2 nucleophiles. Regardless of the nature of the substituent R^1^, R^2^ and R^3^, the (N,N)-spiroketal compounds were obtained in good yield (61–98%) and with high enantioselectivity (77–99%). The lowest enantioselectivity (77%) was observed with the cyclopropyl alkyne-derived substrate. In a subsequent study, Singh and coworkers described a related transformation (Scheme 15b). Using slightly modified conditions, they observed the same yields and the same enantioselectivities with similar alkyl groups [34].
Based on control experiments and mechanistic studies, the authors proposed the following mechanism based on ion-pair and hydrogen-bonding catalysis (Scheme 16). The first step would be the formation of the acyliminium 23. In contrast to Singh’s finding, Sun and Li observed the formation of a covalent phosphate ester intermediate 24 by ^1^H NMR, which would act as a pre-catalyst. This would be followed by a 1,4-addition reaction furnishing tetra-substituted allene 25. Protonation of 25, followed by intramolecular cyclization, would lead to the desired product 22. Interestingly, when 3-unsubstituted 1H-indole is used, the 1,2-addition takes place instead of the 1,4-addition reaction.
The presence of multiple stereogenic elements within a molecule can have a significant impact on its chemical, biological and physical properties. These fascinating molecules are not only encountered in nature, but also designed for a variety of applications, including use as organocatalysts and ligands in enantioselective catalysis. Consequently, molecules bearing multiple stereogenic elements have attracted significant interest, as their distinct three-dimensional features can be exploited in many different areas [35].
In this context, Li, Dong and Li successfully developed an organocatalytic approach for the construction of both axial and central chirality thanks to a similar CPA-catalyzed (3 + 2) annulation between 3-alkynyl-3-hydroxyisoindolinones (α-FPAs) 20 and 1-(3-indolyl)naphthalen-2-ols 21 (Scheme 17a) [36]. This strategy allowed the smooth construction of enantioenriched (N,N)-spiroketal moieties 22, bearing both one stereogenic center and one chirality axis, with excellent diastereo- and enantioselectivities (77–93% yield, 11:1–20:1 dr and 80–97% ee). As previously stated, when R^1^ is a cyclopropyl group, the stereoselectivity is drastically affected, leading to the formation of a racemic product. At the same time, Zhang and Shi reported the same strategy with the same efficiency starting from 3,3-bisindoles 21 as 1,2-dinucleophiles (Scheme 17b) [37].
3.2. Photocatalytic Process
Chiral hydrogen-bond catalysis has been successfully applied to a wide range of stereoselective photocatalytic radical-based reactions. A critical problem for these processes is the formation of secondary anionic intermediates that interact preferentially with the protons of chiral catalysts, thus compromising the enantioselective control. The key innovation brought about by Jiang’s group lies in modulating the redox mechanism (single-electron transfer, SET) towards an energy transfer (EnT) process. This adjustment shifts the generation of anionic intermediates in chiral catalysis by hydrogen bonding to a stage after the formation of stereogenic centers. The result is an enhanced preferential interaction between acid catalysts and key radical species. The researchers have succeeded in carrying out the first stereoselective photochemical spirocyclization reaction for building spiro quaternary stereogenic centers through enantioselective radical addition (Scheme 18) [38]. Through use of a dual-catalysis system that combines a photosensitizer and a chiral Brønsted acid, a wide range of olefinic sulfonyl oximes 26 can be efficiently reacted with vinyl azides 27 to produce azaarene-functionalized (N,N)-spiroketals 28 in good yields (43–94%) and with excellent enantioselectivity (63–99%). The two hydrogen-bonding interactions between the chiral CPA and azarene 29 would be responsible for the enantiocontrol of the 5-endo-trig cyclization step. It is worth noting that the lowest enantioselectivity was observed when R^2^ was a cyclobutyl group. The incorporation of a sulfonyl protecting group on the oximes enables these transformations to be initiated via an EnT process rather than the SET approach used previously [39].
4. (N,S)-Spiroketals
Sulfur-containing heterocyclic frameworks are commonly found in biologically active molecules and are associated with a wide range of pharmacological properties. Among them, benzothiophene and its derivatives have emerged as particularly interesting structural frameworks. Therefore, significant synthetic efforts have been devoted to developing and diversifying the structure of benzothiophene-based compounds. In addition, introducing fluorine into bioactive molecules can result in significant changes to their chemical, physical and biological properties. The fluorine atom uniquely affects the properties of organic molecules due to its blocking effect in metabolic transformations and its ability to mimic enzyme substrates, increasing their lipophilicity and enhancing bioavailability. Consequently, organofluorine compounds have attracted considerable attention in the development of new drugs and agrochemicals.
In this context, several groups have employed trifluoro- or difluoroethyl benzothiophene ketimines 30 in enantioselective (3 + 2) annulation reactions to construct enantioenriched CF_3_- or CF_2_H-embedded spiro-benzothiophenone 33 (Scheme 19). Indeed, in the presence of a base, which, for the purposes of the following examples, is a tertiary amine carried by a bifunctional squaramide or thiourea catalyst, benzothiophene ketimines 30 are transformed into a 1,3-amphiphilic intermediate 32 and can therefore react with a α,β-unsaturated ketone 31 to form (N,S)-spiroketal units 33 with up to four stereogenic centers. This strategy can be associated with either a two-step mechanism or a concerted 1,3-dipolar (3 + 2) cycloaddition reaction.
The stereoselectivity of this reaction can be explained by a dual-activation model of the catalyst, whereby both substrates are simultaneously activated via hydrogen bonds (Scheme 20). The carbanion formed by the catalyst deprotonating benzothiophene ketimines coordinates with the protonated cinchonidin nitrogen atom to form a five-membered ring. At the same time, the two squaramide or thioamide N–H bonds of the catalyst coordinate with the α,β-unsaturated ketone. The chiral catalyst brings the two substrates close together, inducing a highly stereoselective reaction that delivers the desired product.
In 2022, the group of Yan described a squaramide-catalyzed enantioselective (3 + 2) cycloaddition reaction between the N-(2,2)-difluoroethyl benzothiophene ketimine 30 and the 2-oxoindolin-3-ylidene acetate 31 (Scheme 21) [40]. Under optimized conditions, they obtained the desired CF_2_H-containing bispiro(oxindole-pyrrolidine-benzothiophenone)s 33 with four adjacent stereogenic centers, including two adjacent spirocyclic tetra-substituted stereocenters. These bispiro-compounds were formed in a single diastereomer (>20:1), in high yields (70–96%) and typically with excellent enantioselectivities (64–98% ee), whatever the nature of the substituents borne by both substrates. The lowest enantioselectivity (64% ee) was observed when R^2^ = H. In this case, the 2-oxoindolin-3-ylidene acetate 31 is not very soluble and the reaction has to be performed at room temperature.
In 2023, in collaboration with Huang, Yan reported a similar squaramide-catalyzed enantioselective (3 + 2) cycloaddition strategy from trifluoroethyl benzothiophene ketimine 30 (Scheme 22) [41]. The CF_3_-containing bispiro(oxindole-pyrrolidine-benzothiophenone)s were obtained with the same efficiency. However, the nature and position of the substituents on both partners significantly affect the diastereoselectivity (2:1–>20:1 dr).
As part of their ongoing interest in the synthesis of optically pure F-containing pyrrolidines, the same group reported in 2023 on the use of Meldrum’s acid-derived electron-deficient olefins as dipolarophiles in enantioselective (3 + 2) cycloaddition reactions (Scheme 23) [42]. In the presence of a cinchonidine-thiourea-based bifunctional catalyst, this approach produced highly enantioenriched (60–93% ee) α-CF_2_H pyrrolidines embedded in (N,S)-spiroketals with three adjacent stereogenic centers in good yields (50–92%) and excellent diastereoselectivity (10:1–20:1 dr). The lowest enantioselectivity (60%) was obtained when an electron-withdrawing group (R^1^ = Cl) was present on the 1,3-dipole. Additionally, the use of aliphatic chain-substituted Meldrum’s acid affected the diastereoselectivity (10:1 and 12:1 dr) and the yield (50% and 70%) of the reaction.
Enantiopure spiropyrazolones are well recognized for their diverse and valuable biological activities. Building on this, Li, Huang and Yan have recently applied their expertise in organocatalyzed (3 + 2) cycloaddition reactions of 1,3-dipoles derived from N-(2,2-difluoroethyl) benzothiophene ketimines 31 to synthesize optically active dispiro[benzo[b]thiophene-pyrrolidine-pyrazole] derivatives 33 bearing a (N,S) acetal moiety (Scheme 24) [43]. Indeed, the authors demonstrated that unsaturated pyrazolone derivatives 32 were effective partners in this enantioselective (3 + 2) cycloaddition reaction using a bifunctional thiourea catalyst. Cycloadducts, containing two adjacent spiral units, including one (N,S)-ketal moiety and four contiguous stereogenic centers, were obtained in good yields (66–95%) with general diastereoselectivities (82:18–95:5 dr) and with good-to-high enantioselectivities (70–95% ee).
Two other groups have also recently studied this organocatalyzed (3 + 2) cycloaddition reaction in order to produce spiroacetal (N,S) units from benzothiophene ketimines.
In 2023, Yuan’s group used β-trifluoromethyl enones 32 as a dipolarophile in an enantioselective (3 + 2) cycloaddition reaction involving trifluoroethyl benzothiophene ketimines 31 (Scheme 25) [44]. This strategy employed a chiral bifunctional squaramide–tertiary amine catalyst to prepare challenging and potentially bioactive vicinally bis(trifluoromethyl)-substituted (N,S)-spiroketals bearing four contiguous stereogenic centers with excellent stereocontrol (86–99% ee, >20:1 dr).
Finally, Du demonstrated the efficiency with which 2-arylidene-1,3-diones [45,46] react with N-2,2-difluoroethyl benzothiophenone imines in this type of reaction (Scheme 26) [47]. Under mild conditions, the thiourea-catalyzed enantioselective (3 + 2) cycloaddition produced a range of dispiro[benzothiophenone-indandione-pyrrolidine] products with excellent yields (84–98%). It is worth noting that the stereoselectivity is influenced by the nature of the substituents on both substrates. Indeed, the dispiro-derivatives display enantioselectivities of 3–93% ee and diastereomeric ratios ranging from 9:1 to 20:1.
5. Conclusions
In this review, we describe progress in the field of enantioselective synthesis of (N,O)-, (N,N)- and (N,S)-spiroketals. The strategies are mainly based on metallo- and organocatalyzed (4 + 2) and (3 + 2) annulation reactions, Lewis acid-catalyzed cascade reactions and photocatalytic radical-based reactions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1“Spiro compounds” in IUPAC Compendium of Chemical Terminology 5th ed.International Union of Pure and Applied Chemistry Research Triangle Park, NC, USA 2025 Online Version 5.0.0.10.1351/goldbook.S 05881 · doi ↗
- 2Varela M.T. Dias G.G. de Oliveira L.F.N. de Oliveira G.G. Aguiar F.D. Nogueira J.P. Cruz L.R. Dias L.C. Spirocyclic compounds as innovative tools in drug discovery for medicinal chemists Eur. J. Med. Chem.202528711736810.1016/j.ejmech.2025.11736839952099 · doi ↗ · pubmed ↗
- 3Moshnenko N. Kazantsev A. Chupakhin E. Bakulina O. Dar’in D. Synthetic Routes to Approved Drugs Containing a Spirocycle Molecules 202328420910.3390/molecules 2810420937241950 PMC 10223694 · doi ↗ · pubmed ↗
- 4Gilles L. Antoniotti S. Spirocyclic Compounds in Fragrance Chemistry: Synthesis and Olfactory Properties Chem Plus Chem 202287 e 20220022710.1002/cplu.20220022736367229 · doi ↗ · pubmed ↗
- 5Kotha S.B. Deb A.C. Lahiri K. Manivannan E. Selected Synthetic Strategies to Spirocyclics Synthesis 200916519310.1055/s-0028-1083300 · doi ↗
- 6Acosta-Quiroga K. Rojas-Pẽna C. Nerio L.S. Gutíerrez M. Polo-Cuadrado E. Spirocyclic derivatives as antioxidants: A review RSC Adv.202111219262195410.1039/d 1ra 01170 g 35480788 PMC 9034179 · doi ↗ · pubmed ↗
- 7Basavaraja D. Siddalingeshwar V.D. Athira C.S. Aiswarya S. Sreelakshmi V. Ancy A. Sasidhar B.S. Spiro-hetrocycles: Recent advances in biological application and synthetic strategies Tetrahedron 202517313446810.1016/j.tet.2025.134468 · doi ↗
- 8Kamlar M. Urban M. VeselýJ. Enantioselective Synthesis of Spiro Heterocyclic Compounds Using a Combination of Organocatalysis and Transition-Metal Catalysis Chem. Rec.202323 e 20220028410.1002/tcr.20220028436703545 · doi ↗ · pubmed ↗