Bioinspired Electrochemical Cyclization toward the Divergent Synthesis of Mavacurane- and Akuammiline-Type Alkaloids
Eisuke Sato, Tomohiro Nakahama, Yuika Nomura, Koichi Mitsudo, Seiji Suga

TL;DR
A new electrochemical method enables the synthesis of two types of alkaloids from a shared precursor by adjusting redox conditions.
Contribution
A bioinspired electrochemical approach for divergent synthesis of alkaloid frameworks using redox mediator tuning.
Findings
Iodide-mediated electrolysis leads to the formation of the mavacurane core via iodination and cyclization.
Ferrocene-mediated oxidation produces the akuammiline skeleton through radical formation.
Abstract
We report a divergent electrochemical strategy for the bioinspired synthesis of mavacurane- and akuammiline-type alkaloid frameworks from a common indole–malonate precursor. By tuning the redox mediator, selective N–C or C–C bond formation was achieved. Iodide-mediated electrolysis promoted iodination of the malonate carbanion, followed by intramolecular nucleophilic cyclization to furnish the mavacurane core. In contrast, ferrocene-mediated oxidation generated a malonate-centered radical that afforded the akuammiline skeleton.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
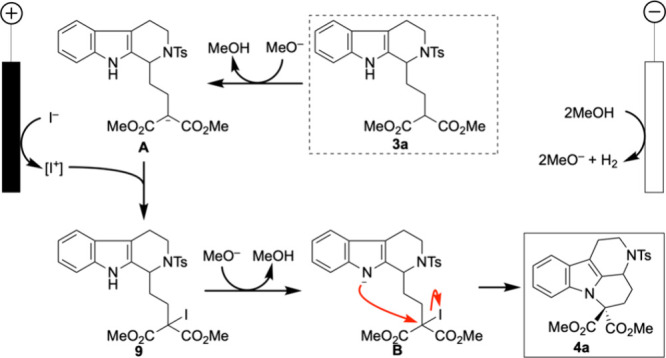 Figure 1
Figure 1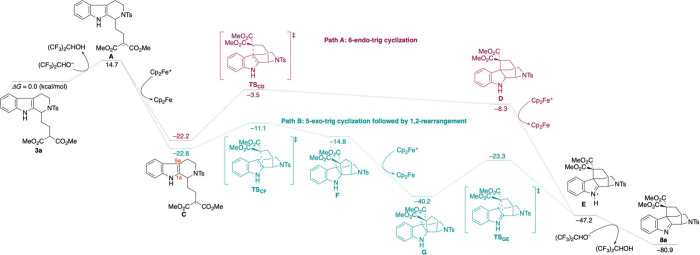 Figure 2
Figure 2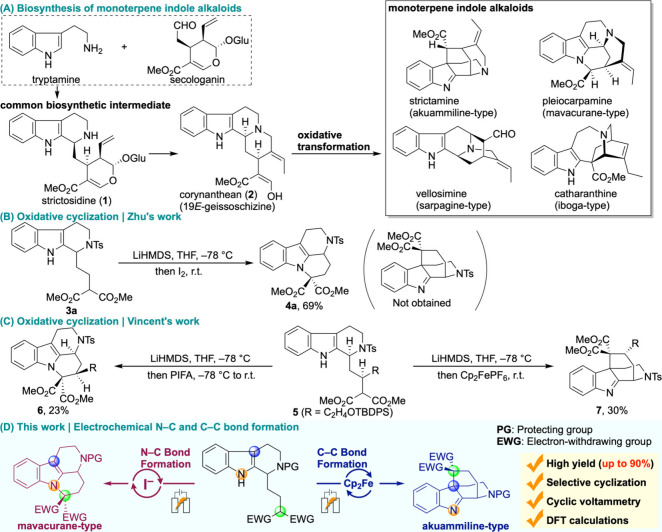 Figure 3
Figure 3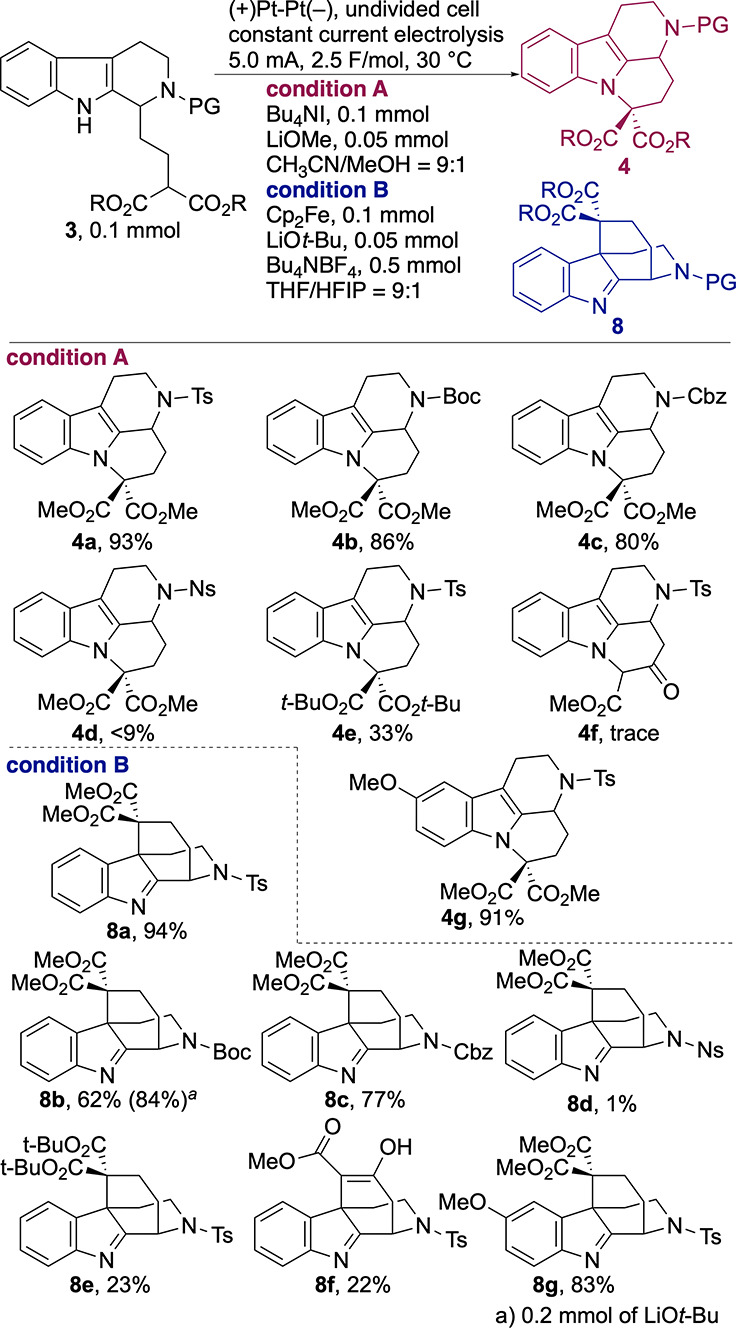 Figure 4
Figure 4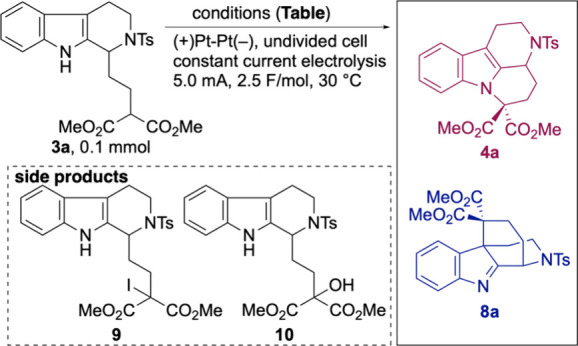 Figure 5
Figure 5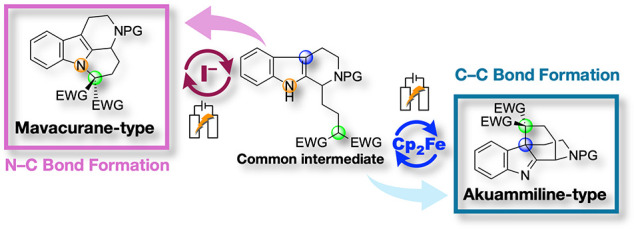 Figure 6
Figure 6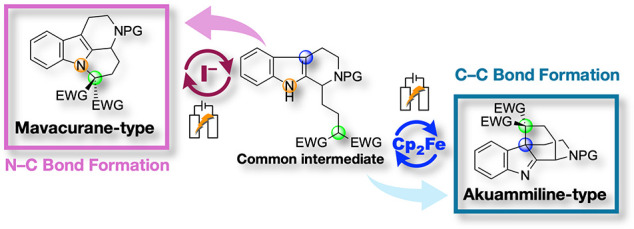 Figure 7
Figure 7- —Asahi Glass Foundation10.13039/100007684
- —Japan Society for the Promotion of Science10.13039/501100001691
- —Japan Society for the Promotion of Science10.13039/501100001691
- —Japan Society for the Promotion of Science10.13039/501100001691
- —Ministry of Education, Culture, Sports, Science and Technology10.13039/501100001700
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAlkaloids: synthesis and pharmacology · Radical Photochemical Reactions · Chemical synthesis and alkaloids
Monoterpene indole alkaloids comprise one of the largest families of natural products, and more than 3000 of these compounds have been isolated from various plant species. ?−? ? These alkaloids exhibit a wide range of structural diversity and biological activities. In biosynthesis, structural diversity is established through the oxidative cyclization of a single biosynthetic intermediate, strictosidine (1).? Following the deglycosylation of 1, a cascade of enzyme-catalyzed transformations, including intramolecular oxidative coupling, gives rise to a vast number of complex scaffolds (SchemeA). These downstream modifications are tightly regulated by enzyme specificity, contributing to the remarkable chemical diversity observed in nature.
Over the past decades, oxidative dianion coupling has been considered a novel tool for the C–C bond formation, and the groups of Baran,? Overman,? and Ma? have developed total syntheses of indole alkaloids through a oxidative strategy. Building on the biosynthetic insight, Zhu reported an oxidative cyclization of a simplified cyclization precursor 3a.? Deprotonation of the 1,3-dicarbonyl moiety followed by I_2_ oxidation induced N–C bond formation, which gave the characteristic mavacurane framework (SchemeB). A subsequent study achieved both N–C and C–C bond formation to access five-membered rings.? Whereas N-iodosuccinimide (NIS) led to N–C bond formation, use of Koser’s reagent (PhI(OH)OTs) promoted C–C bond formation. Vincent later explored a bioinspired oxidative cyclization to construct both the mavacurane and akuammiline scaffolds (SchemeC).? Upon double deprotonation of the malonate and the indole moieties, subsequent addition of iodobenzene bis(trifluoroacetate) (PIFA) favored N–C bond formation, whereas single-electron oxidation with Cp_2_FePF_6_ selectively generated the akuammiline core.
While bioinspired oxidative cyclizations have provided elegant strategies for constructing the architecturally complex skeletons, these methods suffer from significant limitations derived from the reliance on stoichiometric amounts of a strong base and oxidant. To overcome these challenges, we turned to electrochemical oxidation as an alternative activation mode. Electrochemical synthesis offers a tunable platform for redox transformations that proceed without the need for external oxidants or reductants. ?−? ? ? ? This operational simplicity has led to a surge of interest in electrochemical methods for natural product synthesis and complex molecule construction. ?−? ?
Of particular relevance to our study is the concept of indirect electrolysis. ?−? ? This approach offers key advantages in terms of selectivity and functional group tolerance. ?,? A wide array of redox mediatorshalide ions,? ferrocene (Cp_2_Fe) derivatives, ?,? triarylamines? and hypervalent iodines ?,? have been used. Notably, the oxidation behavior of 1,3-dicarbonyl compounds was found to be dependent on the nature of the redox mediator.? Zhang and Huang reported that iodide-mediated electrolysis enables selective N–C bond formation,? while Xu demonstrated that Cp_2_Fe catalyzes the generation of carbon-centered radicals from 1,3-dicarbonyl substrates, enabling C–C bond-forming reactions. ?,? Most recently, Guo developed nickel-catalyzed enantioselective dehydrogenative coupling using Cp_2_Fe as an electrocatalyst.?
Building upon these precedents, we envisioned that electrochemical activation could be harnessed to achieve divergent oxidative cyclizations from a common intermediate, mimicking biosynthetic logic. Specifically, we demonstrate that iodide-mediated electrolysis affords mavacurane frameworks (SchemeD). In contrast, the use of Cp_2_Fe switches the selectivity, enabling access to akuammiline-type frameworks.
We began our investigation by exploring the anodic oxidation of substrate 3a using iodide as a redox mediator (Table). Drawing upon our previous studies ?,? and the precedent established by Zhang and Huang,? the reaction was conducted using Bu_4_NI and LiOMe in CH_3_CN/MeOH as the electrolyte components. Electrolysis with platinum electrodes smoothly delivered desired mavacurane-type product 4a in an excellent 93% yield (entry 1). Substitution with LiI instead of Bu_4_NI afforded 4a in only 20% yield, along with the iodinated side product 9 in 44% yield (entry 2). We next assessed the effect of the halide identity on this cyclization. Use of bromide as the mediator provided 4a in a moderate yield (entry 3), while chloride failed to promote productive cyclization (entry 4). Electricity was essential for the cyclization to proceed, and the control experiment conducted without electricity resulted in the recovery of the starting material (entry 5).
Having established efficient conditions for the construction of the mavacurane framework, we next turned our attention to the synthesis of the akuammiline scaffold. Inspired by Vincent? and Xu’s ?,? work, we carried out the anodic oxidation of 3a using Cp_2_Fe and LiOMe in THF/MeOH (entry 6). Although ^1^H NMR analysis of the reaction mixture confirmed the formation of the desired C–C bond, numerous decomposition products were also observed. We hypothesized that methoxide may nucleophilically attack the C2 or C3 position of indole in the cation intermediates (vide infra) or the imine moiety in 8a. Subsequent degradation of the methanol adduct would lower the isolated yield of 8a.
To address this issue, we investigated the combination of less nucleophilic bases and alcohols. Gratifyingly, LiOt-Bu/t-BuOH afforded 8a in 57% yield (entry 7). Furthermore, replacing t-BuOH with 1,1,1,3,3,3-hexafluoroisopropyl alcohol (HFIP) dramatically improved the yield, affording 8a in 94% yield (entry 8). The radical-stabilization effect of HFIP has been well documented, ?−? ? ? ? ? ? and several radical-mediated transformations have been successfully carried out in HFIP. ?−? ? In our system, both the malonate radical and its intermediate adduct (vide infra) are likely stabilized by HFIP. When the reaction was conducted under an O_2_ atmosphere, the formation of the akuammiline skeleton was strongly inhibited (entry 9). As an alternative, alcohol 10 was obtained in 23% yield with several unidentified compounds. This result supports the generation of a malonate radical, which is likely trapped by O_2_ to form 10. The cyclization also required electricity, and in its absence the starting material was recovered (entry 10).
With the optimized electrochemical conditions in hand, we next explored the substrate scope with respect to nitrogen protecting groups (Scheme). We found that common N-protecting groups were compatible with iodide-mediated N–C bond formation. For instance, replacement of the Ts group with either Boc or Cbz protection did not affect the cyclization efficiency, affording the corresponding products 4b and 4c in 86% and 80% yields, respectively. Similar to iodide-mediated N–C cyclization, both Boc and Cbz groups were well tolerated in place of Ts. Notably, Boc-protected substrate 3b delivered the akuammiline-type product 8b in 62% yield. Since anodic oxidation generates acidic species, which is called electro-generated acid,? we hypothesized that acidic Boc deprotection could diminish the yield. Indeed, the addition of 0.2 mmol of t-BuOLi enhanced the yield of 8b to 84%. The Cbz-protected analogue was also compatible with this reaction and gave 8c in a 77% yield. In contrast, the use of a Ns group significantly hindered the cyclization. The iodide-mediated electrolysis of Ns precursor 3d led to a complex mixture and furnished the desired product 4d in only 9% yield with unidentified impurities. In addition, electrolysis with a Cp_2_Fe mediator also resulted in a highly complex reaction mixture, and only 1% of 8d could be isolated.
The reaction also exhibited a sensitivity to steric hindrance. Substrate 3e, bearing a di-tert-butyl malonate moiety, underwent N–C bond formation to give 4e in 33% yield. The Cp_2_Fe-mediated C–C bond-forming reaction was also sensitive to steric hindrance. The anodic oxidation of 3e significantly decreases the efficiency, affording the desired akuammiline-type product 8e in only a 23% yield.
To further expand the synthetic utility of this methodology, we examined β-ketoester substrate 3f as an alternative to the malonate-type nucleophile. Under the iodide-mediated conditions, 3f failed to undergo oxidation, and the starting material was recovered in 36% yield, suggesting a limitation in oxidizability or nucleophilicity of this substrate class. The Cp_2_Fe-mediated electrolysis provided the corresponding enol-type ketoester 8f in 22% yield.
The electronic property of the indole ring did not significantly affect the cyclization. The presence of a methoxy substituent on the indole did not diminish the yield of the mavacurane-type skeleton, affording 4g in 91% yield. Furthermore, the Cp_2_Fe-mediated cyclization of the same precursor 3g furnished the akuammiline-type compound 8g in 83% yield.
To elucidate the mechanism underlying the anodic cyclization, we conducted cyclic voltammetry (CV) experiments.? The comparison of the voltammograms of substrate 3a, products 4a and 8a, Bu_4_NI, and Cp_2_Fe revealed that either iodide or Cp_2_Fe is preferentially oxidized under the reaction conditions (Figures S8, S15). To determine which moiety of 3athe malonate or the indole unitis more readily oxidized by the anodically generated I^+^ or ferrocenium species, we performed control CV experiments with model compounds. When dimethyl malonate was added to a Bu_4_NI or Cp_2_Fe solution under basic conditions, significant increases in anodic current attributed to I^–^ and Cp_2_Fe were observed (Figures S9, S19). These results indicate that the malonate anion, generated under basic conditions, undergoes indirect oxidation mediated by I^+^ or ferrocenium ions. In contrast, when N-Ts-1,2,3,4-tetrahydro-β-carbolinea model compound for the indole moietywas added to a Bu_4_NI or Cp_2_Fe solution, no appreciable catalytic current was observed (Figures S11, S18). This finding suggests that the indole unit is not directly oxidized by I^+^ or ferrocenium species under these conditions.
Based on the insights gained from cyclic voltammetry and control experiments described in Supporting Information, we propose a reaction mechanism that underlies the formation of the mavacurane-type product 4a (Figure). The sequence begins with deprotonation of the malonate moiety in substrate 3a, generating the anionic intermediate A. The anodic oxidation of iodide generates electrophilic iodine species (I^+^), which are known to react with nucleophiles in solution.? This nucleophilic species undergoes electrophilic iodination, affording iodinated intermediate 9. Subsequently, a second deprotonation step is facilitated by methoxide, and the indole nitrogen attacks the iodinate malonate, leading to N–C bond formation and construction of the mavacurane core to yield 4a. This proposed stepwise mechanism is further supported by DFT calculations (see Supporting Information). Notably, the calculated activation energy for the key cyclization step (from intermediate B to product 4a) is only 1.3 kcal/mol, indicating that the transformation is kinetically accessible.
To gain an understanding of the Cp_2_Fe-mediated C–C bond formation, we performed DFT calculations (Figure). After deprotonation of the malonate moiety, single-electron oxidation of intermediate A by the ferrocenium ion furnishes malonate radical C. The electrophilic nature of malonate radical B allows for cyclization.
We first considered a 6-endo-trig cyclization pathway (Path A), in which the malonate radical adds to the C5a position, forming radical intermediate D. This species could then undergo oxidation to give iminium intermediate E, which upon deprotonation would yield the desired product 8a. While this pathway is a simple explanation for construction of the akuammiline skeleton, DFT calculations revealed a relatively high activation barrier for the C to D transformation (ΔG ^ ‡ ^ = 18.7 kcal/mol), suggesting that this route may not be kinetically favorable.
We therefore considered an alternative and more favorable pathway: a 5-exo-trig cyclization, followed by a 1,2-rearrangement (Path B). In this scenario, the malonate radical C undergoes 5-exo-trig cyclization to form the benzylic radical F, proceeding via a significantly lower activation barrier (ΔG ^ ‡ ^ = 11.7 kcal/mol). Oxidation of F affords benzylic cation G. Semipinacol rearrangement generates the same iminium species E, which is subsequently deprotonated to furnish compound 8a.
Importantly, all calculated energy barriers associated with Path B were markedly lower than those for Path A. These findings strongly support the conclusion that the anodic cyclization route to the akuammiline-type skeleton most likely proceeds via the 5-exo-trig cyclization/1,2-rearrangement pathway.
In summary, we have developed an electrochemical cyclization strategy to access both mavacurane- and akuammiline-type skeletons. A common intermediate bearing an indole core and a malonate moiety could be selectively oxidized by using either iodide or Cp_2_Fe as a redox mediator. The choice of mediator dramatically influenced the cyclization pathway, enabling the selective formation of both frameworks in high yields.?
Overall, our findings highlight the potential of electrochemical control in guiding complex skeletal transformations and pave the way for the rational design of oxidative cyclization strategies for alkaloid synthesis. Further studies toward the total synthesis of natural products using this electrochemical methodology are currently underway in our laboratory.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1O’Connor S. E.Maresh J. J.Chemistry and biology of monoterpeneindole alkaloid biosynthesis Nat. Prod. Rep.20062353210.1039/b 512615 k 16874388 · doi ↗ · pubmed ↗
- 2Umer S. M.Solangi M.Khan K. M.Saleem R. S. Z.Indole-Containing Natural Products 2019–2022: Isolations, Reappraisals, Syntheses, and Biological Activities Molecules 202227758610.3390/molecules 2721758636364413 PMC 9655573 · doi ↗ · pubmed ↗
- 3Salim V.Jarecki S.-A.Vick M.Miller R.Advances in Metabolic Engineering of Plant Monoterpene Indole Alkaloids Biology 202312105610.3390/biology 1208105637626942 PMC 10452178 · doi ↗ · pubmed ↗
- 4Baran P. S.Richter J. M.Direct Coupling of Indoles with Carbonyl Compounds: Short, Enantioselective, Gram-Scale Synthetic Entry into the Hapalindole and Fischerindole Alkaloid Families J. Am. Chem. Soc.20041267450745110.1021/ja 047874 w 15198586 · doi ↗ · pubmed ↗
- 5Martin C. L.Overman L. E.Rohde J. M.Total Synthesis of (±)-Actinophyllic Acid J. Am. Chem. Soc.20081307568756910.1021/ja 803158 y 18491907 PMC 2654095 · doi ↗ · pubmed ↗
- 6Zuo Z.Xie W.Ma D.Total Synthesis and Absolute Stereochemical Assignment of (−)-Communesin FJ. Am. Chem. Soc.2010132132261322810.1021/ja 106739 g 20812683 · doi ↗ · pubmed ↗
- 7Ren W.Tappin N.Wang Q.Zhu J.Synthetic Study towards Strictamine: The Oxidative Coupling Approach Synlett 2013241941194410.1055/s-0033-1339472 · doi ↗
- 8Andres R.Wang Q.Zhu J.Asymmetric Total Synthesis of (−)-Arborisidine and (−)-19-epi-Arborisidine Enabled by a Catalytic Enantioselective Pictet–Spengler Reaction J. Am. Chem. Soc.2020142142761428510.1021/jacs.0c 0580432692169 · doi ↗ · pubmed ↗