B/T Mixed Phenotype Acute Leukaemia Harbouring NUP98::BPTF Fusion
Van Tuong Nguyen, Lina Han, Jing Xu, Miguel Cantu, Franklin Fuda, Samuel John, Tamra Slone, Weina Chen

TL;DR
A rare case of mixed B/T acute leukemia with a NUP98::BPTF fusion is reported, offering new insights into its genetic features and treatment challenges.
Contribution
This is the first case study of B/T mixed phenotype acute leukemia with NUP98::BPTF fusion and T-lineage characteristics.
Findings
The patient had a t(11;17)(p15;q23)/NUP98::BPTF fusion and mutations in NOTCH1, FBXW7, and PHF6.
The patient failed initial T-ALL-directed chemotherapy but achieved remission after consolidation therapy.
This case provides new clinicopathological and genomic insights into NUP98::BPTF-rearranged leukemia.
Abstract
NUP98::BPTF rearranged (r) leukaemia is rare with only five reported cases, including acute myeloid leukemia and T‐lymphoblastic leukemia (T‐ALL). Herein, we report a case of NUP98::BPTF‐r mixed phenotype acute leukemia (MPAL), B/T with T‐lineage‐predominance in a 15‐year‐old female. Cytogenetic and molecular studies revealed a t(11;17)(p15;q23)/NUP98::BPTF fusion and cooperative gene alterations characteristic of T‐ALL (NOTCH1, FBXW7, and PHF6). Our patient had a primary induction failure with T‐ALL‐directed chemotherapy and achieved complete remission following consolidation. This is the first case study characterizing the clinicopathological and genomic features of B/T MPAL harboring NUP98::BPTF fusion and providing insights into molecular pathogenesis. The authors have confirmed clinical trial registration is not needed for this submission.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
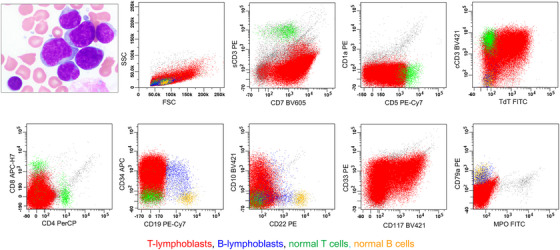 FIGURE 1
FIGURE 1| Case (References) | Age/Sex | Cytogenetics | Other gene alterations | Chemotherapy | HSCT | A/D (follow‐up months) |
|---|---|---|---|---|---|---|
| AMML [ | 10 month/M | 46, XY, t(11;17) (p15;q23)[20] | NA | Anthracycline/ cytarabine/ etoposide | Yes |
A/(51) |
| AMKL [ | 8 month/M | 49, XY, +6, +7, t(11;17)(p15;q23), +19 |
NA |
Induction: low‐dose cytarabine, daunorubicin, etoposide, followed by Intensification and TVTC15 | NA | A/(NA) |
| AML [ | 20 year/F | 46, XX, del(5)(q31), t(11;12)(p13;p13) [4] / 45, idem, ‐13 [5] / 46, XX [1] |
|
Induction 3+7 |
NA |
NA |
| T‐ALL [ | 19 year/F | 86∼89, XXXX, +X, add(1)(p36), ‐4, ‐ 5, 6, ?add(6)(q21), ‐7, ‐8, ‐9, ‐10, ‐11, der(11)add(11)(p11) add(11)(q23), ‐13, ‐14, ‐15, ‐16, ‐17, ‐18, ‐18, ‐20, ‐21, +11, ∼15mar, inc [cp5] / 46, XX [14] |
|
ALL202‐U |
Yes |
A (NA, CR) |
| AML [ | 0‐3/M | 47, XY, +6, t(11;17)(p15;q23)[2]/48, idem, +6[10]/49, idem, +6, +21[2]/46, XY[6] | RNAseq: cluster in AMKL |
NA |
NA |
NA |
| MPAL, T/B‐T (our patient) |
15 year/F | 46, XX, t(11;17)(p15;q23)[6]/47, idem, +4[12]/46, XX[2] | DNAseq: | InductionAALL1231 |
No | A/(4.3, CR |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Myeloid Leukemia Research · Acute Lymphoblastic Leukemia research · Virus-based gene therapy research
Introduction
1
Mixed phenotype acute leukemia (MPAL) is an aggressive leukemia in which leukemia blasts exhibit immunophenotypic evidence of differentiation along more than one hematopoietic cell lineage [1, 2]. Within this category, B/T MPAL is uncommon, accounting for only ∼5% of MPAL cases, and characterized by blasts expressing both B‐ and T‐lineage defining markers. Diagnosis and clinical management of MPAL are often challenging due to its rarity, immunophenotypic and genomic complexity, and lack of biologically informed treatment guidelines [3].
The NUP98 gene (chromosome 11p15) encodes a nucleoporin protein that plays a role in molecular transport across the nuclear envelope. The NUP98 fusions involve the N‐terminus of the NUP98 protein and the C‐terminus of a partner (including HOX genes and non‐HOX genes) and result in impairing differentiation and enhancing self‐renewal of hematopoietic stem cells [4].
The NUP98 fusions are present in ∼5% of childhood acute leukemia, most frequently as NUP98::NSD1 in acute myeloid leukemia (AML), NUP98::KDM5A in acute megakaryoblastic leukemia (AMKL), and less commonly NUP98::RAP1GDS1 and NUP98::KDM5A in T‐lymphoblastic leukemia (T‐ALL) [4, 5]. The NUP98 fusions have been recently reported in rare cases of MPAL, including NUP98::NSD1 in B/Myeloid (M) MPAL, NUP98::MLLT1 in B/T MPAL, and NUP98:IQCG in T/M MPAL [6, 7, 8]. These leukemias are considered a high‐risk subtype of leukemia with poor prognosis, including induction failure [4].
Herein, we present the first case of B/T MPAL with T‐lineage predominance and harboring NUP98::BPTF fusion in a pediatric patient. This fusion is extremely rare with only five reported cases in the literature [5, 9, 10, 11, 12], spanning diverse phenotypes—AML [including AMKL and AMML (acute myelomonocytic leukemia)] and T‐ALL. This case study characterizes the clinicopathological features and cooperative gene alterations of B/T MPAL harboring NUP98::BPTF fusion and provides insights into leukemogenesis.
Result (Case Description)
2
A 15‐year‐old female with a history of scoliosis presented with two weeks of intermittent fever and worsening fatigue. Physical examination was unremarkable except for signs of anemia; no significant lymphadenopathy or organomegaly was noted. Laboratory evaluation revealed pancytopenia with a white blood cell count of 2.3 × 10^3^/µL with 45% blasts, hemoglobin of 3.0 g/dL, and platelet count of 56 × 10^3^/µL. Bone marrow (BM) examination revealed a hypercellular marrow with nearly complete replacement by medium‐sized to large blasts with moderately condensed chromatin, irregular nuclei, small or inconspicuous nucleoli, and scant cytoplasm (Figure 1). Flow cytometric analysis (BD‐Cantor, 10‐color panel [13]) identified two CD34(+)/TdT(+) neoplastic populations of lymphoblasts with a predominant population of T‐lymphoblasts (92% of the total events) that demonstrated characteristics of early T‐cell progenitor T‐ALL (ETP‐ALL), including expression of cytoplasmic CD3 (strong), CD117, and CD5 (partial) but absent surface CD3 and CD1a, and a separate minor population of B‐lymphoblasts (0.8% of the total events) (Figure 1). The immunophenotypic profile met the fifth edition of the World Health Organization Classification of Tumors of the Hematopoietic and Lymphoid Tissues (WHO‐HEM5) diagnostic criteria for MPAL, B/T subtype with a T‐lineage predominance, which permits the diagnosis of MPAL regardless of the percentage of the individual neoplastic blast population [1].
B/T MPAL (bilineal, consisting of T‐lymphoblasts and B‐lymphoblasts with T‐lymphoblast predominance). The upper left image demonstrates the morphologic features of lymphoblasts in BM aspirate: medium‐sized to large lymphoblasts with moderately condensed chromatin, irregular nuclei, small or inconspicuous nucleoli, and scant cytoplasm. By flow cytometric immunophenotyping (10‐color BD Canto and a panel of lymphoid/myelomonocytic markers [13]), there are two neoplastic populations of lymphoblasts, T‐lymphoblasts and B‐lymphoblasts. T‐lymphoblasts (in red) are predominant (92% of the total events), medium to large in size [with increased forward scatter (FSC)], and express cytoplasmic CD3 (cCD3), CD5 (partial), CD10(partial), CD7, CD33, CD34, CD117, and TdT, but lack expression of CD1a, surface CD3 (sCD3), CD4, CD8, CD19, or myeloperoxidase (MPO). This immunophenotype is consistent with ETP‐ALL (early T‐progenitor T‐ALL). B‐lymphoblasts (in blue) represent a minor population (0.8% of the total events), are medium‐sized (with low FSC), and express CD10 (a small subset), CD19, CD22 (partial), CD79a, CD34, and TdT (partial), but lack expression of sCD3, cCD3, or MPO.
Conventional karyotyping revealed a t(11;17)(p15;q23) that resulted in the NUP98::BPTF fusion, confirmed by next‐generation sequencing (NGS by DNAseq and RNAseq on a BM sample), and additional abnormalities (second clone of 47, idem, +4). NGS also identified other gene alterations in NOTCH1 (p.Leu1585Gln, VAF 27.61%), NOTCH1 (p.Leu1678Pro, VAF 16.76%), FBXW7 (p.Arg689Trp, VAF 22.88%), PHF6 (p.Arg274Gln, VAF 48.21%), and SH2B3 (p.Pro432fs, VAF 27.80%) (Table 1). No rearrangements of KMT2A, CRLF2, or BCR::ABL1 were present.
Given the T‐lineage predominance with only a minor population of B lymphoblasts, the patient was treated with T‐ALL‐directed induction chemotherapy (COG AALL1231) but showed refractory disease on day 29 BM with 60% T/B lymphoblasts with T‐lineage predominance. She was proceeded with consolidation chemotherapy per COG AALL0434 with nelarabine that led to measurable residual disease (MRD, 0.01% sensitivity) negative remission (4.3 months since diagnosis) with a plan to proceed with additional chemotherapy.
Discussion
3
This case study reports the first case of B/T MPAL harboring the NUP98::BPTF fusion and cooperative gene alterations, thus expanding the existing phenotypic spectrum of NUP98::BPTF rearranged leukemia (AML and T‐ALL) to include B/T MPAL. All six patients (5 patients reported previously [5, 9, 10, 11, 12] and our patient) were young with a median age of 8.3 years and male to female ratio of 1 and presented with various leukemia phenotypes, AML (including AMML and AMKL) in most patients (4/6), T‐ALL and B/T MPAL each in one patient (1/6). All patients had abnormal cytogenetic abnormalities, including most patients (4/6) with complex karyotype. Furthermore, NUP98::BPTF fusion was cytogenetically evident in most patients (4/6) by the presence of t(11;17)(p15;q23). Of four patients with clinical follow‐up, all patients (including our B/T‐T MPAL) exhibited an aggressive clinical course initially, including primary refractory disease or early relapse, consistent with the known association of NUP98‐rearranged leukemia with chemoresistance [4]. Three patients were proceeded with allogeneic hematopoietic stem cell transplant (allo‐HSCT) that led to a durable remission; our patient has a plan to receive HSCT. These four patients were alive at the last follow‐up.
The NUP98::BPTF fusion encodes a chimeric protein combining the FG‐repeat domains of NUP98 with the PHD finger and bromodomain of BPTF. Alternative splicing yields variants with one or two PHD domains, and its expression is associated with activation of HOXA9 and HOXA10, as well as upregulation of the proto‐oncogene PIM1 [9, 11]. Structurally, it is similar to NUP98::KDM5A, as both fusions retain the FG‐repeat motifs of NUP98 and the C‐terminal chromatin recognition modules of their partner proteins.
While NUP98::BPTF displays significant oncogenic potential by altering transcription and chromatin regulation, current evidence suggests that NUP98 fusions are rarely sufficient on their own to induce leukemia [4]. Experimental models and clinical observations indicate that additional gene alterations, including FLT3, NRAS, KRAS, KIT, or WT1 mutations, are typically required to provide the necessary proliferative or survival advantage [4]. This notion aligns with clinical observations across NUP98‐fusion leukemia, which almost invariably harbor cooperative gene alterations. Indeed, our case of NUP98::BPTF fusion positive B/T MPAL had cooperative gene mutations characteristic of T‐ALL (NOTCH1, FBXW7, and PHF6). Similarly, a case of NUP98::BPTF fusion positive AML had gene expression pattern clustered in AMKL by RNAseq [5]. These findings are similar to NUP98::KDM5A fusion positive leukemia, such as concurrent NOTCH1 mutation in T‐ALL and RB1 loss in AMKL [4, 5]. Collectively, these findings provide insight into leukemogenesis, fusion oncoproteins and cooperating mutations defining disease phenotypes.
Furthermore, our case demonstrates the recently described phenotype‐genotype association, i.e., the genotype of MPAL being associated with immunophenotypic lineage predominance [8], as seen in our case of B/T MPAL with T‐lineage predominance and cooperative gene alterations characteristic of T‐ALL. This association has clinical implications on the selection of appropriate induction therapy in MPAL, B‐ALL‐, T‐ALL‐, or AML‐based regimen [13]. Our patient was initially treated with T‐lineage‐matched induction therapy but experienced induction failure and achieved remission following consolidation chemotherapy (COG AALL0434 with nelarabine). This reflects the aggressiveness of NUP98 rearranged leukemias and urges the need for novel therapy targeting NUP98 fusion. Molecular dependency of KMT2A has been demonstrated in NUP98 fusion protein‐driven leukemogenesis [5]. Menin inhibitors have shown anti‐leukemia efficacy in KMT2A‐rearranged leukemia [15]. The efficacy of menin inhibitors remains to be explored in leukemia with NUP98::BPTF fusion protein.
From diagnostic perspective, our case illustrates a discrepancy between the WHO‐HEM5 and the International Consensus Classification (ICC). While this case met the diagnostic criteria of B/T‐T MPAL, it would be classified as T‐ALL with a comment on the minor population of B‐lymphoblasts per the ICC guideline that requires the smaller neoplastic blast population to be at least 5% of the total cells [1, 2]. Whether using the ICC or the WHO‐HEM5 criteria, it is essential to report a minor population of leukemic blasts from a divergent lineage, as this population may become predominant after therapy and have significant therapeutic and prognostic implications [16, 17]. Although optimal induction chemotherapy has not yet been well established in B/T MPAL, current guidelines recommend the ALL‐type induction regimen, most often guided by the dominant immunophenotype [3, 18]; our patient was effectively treated following this guideline as described above.
In summary, this is the first case study that characterizes the clinicopathologic and genomic features of B/T MPAL with NUP98::BPTF and provides insights into leukemogenesis. Future collaborative studies are necessary to better understand leukemia pathogenesis and to develop biologically guided therapies.
Author Contributions
VN, LH, and WC conceptualised the case study and wrote the original draft. All authors critically revised the manuscript and approved the final version of the manuscript.
Ethics Statement
All procedures performed in this case study were part of the clinical management in accordance with the ethical standards.
Consent
Waived per approved protocol (IRB STU 122013–023).
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1J. D. Khoury , E. Solary , O. Abla , et al., “The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms,” Leukemia 36, no. 7 (2022): 1703–1719, 10.1038/s 41375-022-01613-1.35732831 PMC 9252913 · doi ↗ · pubmed ↗
- 2O. K. Weinberg , D. A. Arber , H. Dohner , et al., “The International Consensus Classification of Acute Leukemias of Ambiguous Lineage,” Blood 141, no. 18 (2023): 2275–2277.36877915 10.1182/blood.2022019493 · doi ↗ · pubmed ↗
- 3E. Orgel , T. B. Alexander , B. L. Wood , et al., “Mixed‐Phenotype Acute Leukemia: A Cohort and Consensus Research Strategy From the Children's Oncology Group Acute Leukemia of Ambiguous Lineage Task Force,” Cancer 126, no. 3 (2020): 593–601, 10.1002/cncr.32552.31661160 PMC 7489437 · doi ↗ · pubmed ↗
- 4N. L. Michmerhuizen , J. M. Klco , and C. G. Mullighan , “Mechanistic Insights and Potential Therapeutic Approaches for NUP 98‐Rearranged Hematologic Malignancies,” Blood 136, no. 20 (2020): 2275–2289, 10.1182/blood.2020007093.32766874 PMC 7702474 · doi ↗ · pubmed ↗
- 5M. Umeda , R. Hiltenbrand , N. L. Michmerhuizen , et al., “Fusion Oncoproteins and Cooperating Mutations Define Disease Phenotypes in NUP 98‐Rearranged Leukemia,” Blood (2025): blood‐2025028993.10.1182/blood.202502899340700635 · doi ↗ · pubmed ↗
- 6Q. Pan , Y.‐J. Zhu , B.‐W. Gu , et al., “A New Fusion Gene NUP 98‐IQCG Identified in an Acute T‐Lymphoid/Myeloid Leukemia With a t(3;11)(q 29q 13;p 15)del(3)(q 29) Translocation,” Oncogene 27, no. 24 (2008): 3414–3423, 10.1038/sj.onc.1210999.18084320 · doi ↗ · pubmed ↗
- 7W. Xiao , M. Bharadwaj , M. Levine , et al., “PHF 6 and DNMT 3A Mutations Are Enriched in Distinct Subgroups of Mixed Phenotype Acute Leukemia With T‐Lineage Differentiation,” Blood Advances 2, no. 23 (2018): 3526–3539, 10.1182/bloodadvances.2018023531.30530780 PMC 6290101 · doi ↗ · pubmed ↗
- 8R. Zheng , F. Fuda , J. R. Gagan , et al., “Genomic Heterogeneity Within B/T Mixed Phenotype Acute Leukemia in a Context of an Immunophenotype,” Leukemia Research Reports 21 (2024): 100410.38273970 10.1016/j.lrr.2023.100410 PMC 10808966 · doi ↗ · pubmed ↗