Genomic characterization of Citrobacter freundii clinical isolates from Tanzania
Gerald Mboowa, Benson R. Kidenya, Ivan Sserwadda, Stephen Kanyerezi, Inyasi Lawrence Akaro, Baraka Mkinze, Jeremiah Seni

TL;DR
This study analyzes the genomes of three Citrobacter freundii isolates from Tanzania, revealing antibiotic resistance and virulence traits.
Contribution
The novelty lies in genomic characterization of C. freundii isolates from Tanzania, revealing multidrug resistance and virulence factors.
Findings
Draft genomes of three C. freundii isolates were sequenced with an average of 6.9 Mb and 51.49% GC content.
ST167 isolate showed multiple resistance genes and virulence factors, indicating a multidrug-resistant lineage.
The findings emphasize the importance of genomic surveillance for tracking emerging C. freundii strains.
Abstract
We report draft genomes of three Citrobacter freundii isolates from Tanzania. Genomes averaged 6.9 Mb with 51.49% GC content and 114× depth. ST167 carried multiple extended-spectrum β-lactamase, fluoroquinolone, and aminoglycoside resistance genes alongside virulence factors, highlighting the need for genomic surveillance of emerging multidrug-resistant C. freundii lineages.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
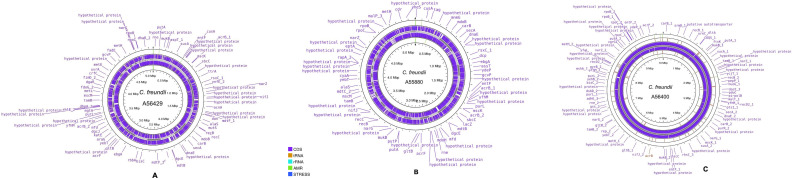 Fig 1
Fig 1| Strain | A55880 | A56400 | |
|---|---|---|---|
| ST | ST116 | ST167 | ST263 |
| Genome size (bp) | 5,502,433 | 10,239,272 | 5,160,700 |
| G + C (%) | 51.61 | 51.18 | 51.67 |
| Number of contigs | 144 | 225 | 59 |
| Size of the largest contig | 503,328 | 273,666 | 836,533 |
| N50 (bp) | 173,136 | 13,036 | 276,703 |
| Completeness | |||
| Sequencing depth | 133× | 62× | 147× |
| CDS (total) | 5,228 | 9,484 | 4,825 |
| Number of tRNAs | 309 | 631 | 309 |
| Drug resistance genes |
|
|
|
| Plasmid replicons |
|
|
|
| Virulence and stress genes |
|
|
|
| SRA accession number |
|
|
|
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Genomics and Phylogenetic Studies · Enterobacteriaceae and Cronobacter Research
ANNOUNCEMENT
Citrobacter freundii is a Gram-negative bacterium from the Enterobacteriaceae family, typically considered part of the human gut flora but increasingly implicated in healthcare-associated infections, particularly among immunocompromised patients (1). The emergence of multidrug-resistant strains and the acquisition of resistance genes via mobile genetic elements have heightened concern for infection control and antimicrobial stewardship programs.
As part of a genomic surveillance study, we investigated C. freundii isolates collected between January and May 2020 from orthopedic wards at Bugando Medical Centre, a 900-bed tertiary referral hospital in Mwanza, Tanzania. The isolates were part of a broader study aimed at evaluating the transmission of extended-spectrum β-lactamase (ESBL)-producing bacteria in clinical and hospital environments. A total of 283 participants were enrolled, including 265 index patients and 18 neighboring patients. Additional samples were collected from 18 non-medical caregivers, 24 healthcare workers, and 88 environmental surfaces (2).
Sample collection involved rectal or stool swabs from patients within 24 hours of admission. ESBL-producing Gram-negative bacteria were identified via selective culture on MacConkey agar supplemented with cefotaxime (2). Bacterial identification was done using standard biochemical tests, including oxidase, catalase, citrate utilization, and triple sugar iron agar reactions. Antimicrobial susceptibility testing was performed according to Clinical and Laboratory Standards Institute (CLSI) guidelines using the Kirby-Bauer disk diffusion method. The antibiotic panel included cefotaxime, ceftazidime, ciprofloxacin, gentamicin, meropenem, and trimethoprim-sulfamethoxazole. Confirmation of ESBL production was done using the combined disk method (2).
After presumptive ESBL-producing Gram-negative bacteria were identified on MacConkey agar with cefotaxime, the cultures were incubated aerobically at 35–37°C for 18–24 hours. Colonies with distinct morphology were subcultured for a purity check. Confirmed Enterobacteriaceae isolates underwent genomic DNA extraction using the QIAamp DNA Mini Kit (Qiagen, Germany) as per the manufacturer’s instructions. DNA quality and quantity were assessed using NanoDrop 2000 spectrophotometry (Thermo Fisher Scientific, USA). Subsequently, whole-genome sequencing was performed at the Earlham Institute (Norwich, UK) using the Low Input Transposase Enabled Illumina library preparation protocol on the NovaSeq 6000 platform, generating 150 bp paired-end reads.
Bioinformatic analysis was performed using the rMAP pipeline v1.0 (3) for species genomic confirmation, antimicrobial resistance (AMR) gene detection, MLST typing, and genome annotation. To validate and extend these analyses, the TheiaProk_Illumina_PE_PHB workflow v1.0.0 (4) on the Terra platform was used for quality control assessment, including QUAST v5.2.0 (5) and BUSCO v5.8.2 (6) evaluations, providing robust and reproducible genomic characterizations (Table 1). Quality control was performed with FastQC v0.11.9 (7) and Trimmomatic v0.39 (8). De novo genome assembly was performed using SPAdes v3.15.2 with default parameters optimized for Illumina paired-end data. SPAdes v4.2.0 (9) constructs the assembly using a de Bruijn graph approach and includes read error correction and scaffolding steps, making it well-suited for bacterial genomes. Genome annotation was performed using Prokka v1.14.6 (10), which enabled rapid functional annotation of assembled genomes, assigning gene functions and identifying coding sequences, rRNAs, and tRNAs.
AMR genes were identified using AMRFinderPlus v3.12.8 (11) and ResFinder v 4.7.2 (12) with minimum identity and coverage thresholds set at 90% and 60%, respectively. Plasmid replicons were detected using PlasmidFinder v2.1.1 (13) with default parameters. MLST was determined via the PubMLST database using mlst v2.22.0(14).
Three C. freundii isolates (A55880, A56400, and A56429) were sequenced with an average depth of 114× and genome sizes ranging from 5.1 to 10.2 Mbp (Fig. 1A through C). The unusually large genome size (~10.2 Mb) of C. freundii A56400 may be attributed to the presence of multiple plasmids, integrated prophages, and horizontally acquired mobile genetic elements. GC content ~51.5%. MLST analysis identified the isolates as ST116, ST167, and ST263. ST167 (A56400) exhibited the broadest AMR profile, including blaCTX-M-15, blaTEM-1, blaCMY, qnrB, and aac(6′)-Ib-cr, alongside virulence genes espX1, ybtQ, and ybtP (Table 1). These findings indicate iron acquisition capacity and potential immune evasion via Type III secretion systems. ST263 and ST116 demonstrated moderate and intermediate profiles, respectively, with varying plasmid content. The most common plasmid replicons included IncFIB, IncHI1B, and IncR.
(A–C) Genome circularization of draft C. freundii A56429, C. freundii A55880, and C. freundii A56400. The concentric rings display annotated genomic features, with the outermost ring showing coding sequences (CDS) and genes, including hypothetical proteins, annotated by Prokka on both strands. Functional elements are color-coded: CDS (purple), tRNA (orange), rRNA (cyan), AMR genes (green), and stress response genes (blue). Visualization was performed using Proksee (https://proksee.ca/). Genomic coordinates are marked in megabase pairs (Mbp) around the circle.
This study highlights the genomic diversity and resistance potential of C. freundii strains circulating in Tanzanian healthcare settings. The identification of ST167 as a high-risk clone, harboring diverse AMR and virulence determinants, underscores the need for continued genomic surveillance. These results provide critical insights into infection control strategies in resource-limited settings.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jabeen I, Islam S, Hassan A, Tasnim Z, Shuvo SR. 2023. A brief insight into Citrobacter species - a growing threat to public health. Front Antibiot 2:1276982. doi:10.3389/frabi.2023.127698239816660 PMC 11731968 · doi ↗ · pubmed ↗
- 2Seni J, Akaro IL, Mkinze B, Kashinje Z, Benard M, Mboowa G, Aruhomukama D, Sserwadda I, Joloba ML, Mshana SE, Kidenya BR. 2021. Gastrointestinal tract colonization rate of extended-spectrum beta-lactamase-producing gram-negative bacteria and associated factors among orthopaedic patients in a tertiary hospital in Tanzania: implications for infection prevention. Infect Drug Resist 14:1733–1745. doi:10.2147/IDR.S 30386034007192 PMC 8123940 · doi ↗ · pubmed ↗
- 3Sserwadda I, Mboowa G. 2021. r MAP: the rapid microbial analysis pipeline for ESKAPE bacterial group whole-genome sequence data. Microb Genom 7:000583. doi:10.1099/mgen.0.00058334110280 PMC 8461470 · doi ↗ · pubmed ↗
- 42025. Dockstore | Workflow | Github.Com/Theiagen/Public_health_bioinformatics/Theia Prok_Illumina_PE_PHB. Available from: https://dockstore.org/workflows/github.com/theiagen/public_health_bioinformatics/Theia Prok_Illumina_PE_PHB:v 1.0.0?tab=info
- 5Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi:10.1093/bioinformatics/btt 08623422339 PMC 3624806 · doi ↗ · pubmed ↗
- 6Manni M, Berkeley MR, Seppey M, Zdobnov EM. 2021. BUSCO: Assessing genomic data quality and beyond. Curr Protoc 1:e 323. doi:10.1002/cpz 1.32334936221 · doi ↗ · pubmed ↗
- 7Babraham Bioinformatics. 2025. Fast QC A quality control tool for high throughput sequence data. Available from: https://www.bioinformatics.babraham.ac.uk/projects/fastqc
- 8Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi:10.1093/bioinformatics/btu 17024695404 PMC 4103590 · doi ↗ · pubmed ↗