Two cases of congenital isolated adrenocorticotropic hormone deficiency due to pathogenic variants in <i>TBX19</i>
Ю. Л. Скородок, А. В. Кожевникова, Е. В. Плотникова, И. Ю. Иоффе, А. Н. Тюльпаков

TL;DR
This paper describes two cases of a rare genetic disorder causing adrenal hormone deficiency due to mutations in the TBX19 gene, with detailed clinical and genetic findings.
Contribution
The study reports two new cases of TBX19-related disease, including a previously unreported genetic variant.
Findings
Two patients with congenital isolated ACTH deficiency were found to have pathogenic variants in TBX19.
One patient had a novel variant in TBX19, c.469-1G>A, not previously described in the literature.
Clinical features included hypoglycemia and liver dysfunction, which improved with hydrocortisone therapy.
Abstract
Врожденный изолированный дефицит адренокортикотропного гормона (ВИДА) — орфанное аутосомно-рецессивное заболевание, обусловленное патогенными вариантами в гене ТВХ19 (1q24.2). В статье представлено описание двух клинических случаев с классической манифестацией ВИДА в неонатальном периоде, подтвержденного генетически, причем в одном из них выявлен впервые описанный вариант в гене TBX19. Диагноз установлен на 8-м и 22-м месяцах жизни, несмотря на появление клинически значимых симптомов в периоде новорожденности у обеих пациенток. Клинические проявления гипогликемии присутствовали у обеих пациенток: у пациентки №2 — с первых суток жизни (эпизод апноэ), у пациентки №1 — с 7 месяцев (судороги). У пациентки №1 основными проявлениями заболевания были холестатическая желтуха, гепатомегалия, признаки цитолиза гепатоцитов, нарушение белковосинтетической функции печени, что может свидетельствовать…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
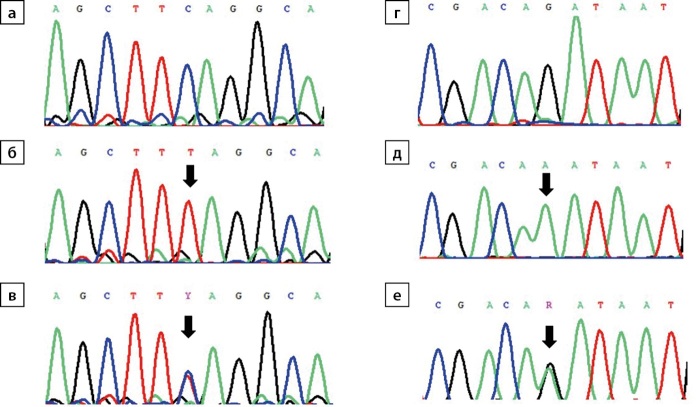 Figure 1
Figure 1| Показатель | Результат | Норма |
| ТТГ, мМЕ/л | 3,52 | 0,62–8,0 |
| свТ4, пмоль/л | 12,9 | 10–26 |
| СТГ, нг/мл | 3,5 | 1,3–9,1 |
| ИФР-1, нг/мл | 29,2 | 28–131 |
| Инсулин, мкЕд/мл | 0,2 | 2,3–26 |
| С-пептид, нг/мл | 0,1 | 0,36–3,6 |
| Глюкоза, ммоль/л | 0,6 | 3,3–5,5 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital heart defects research · Digestive system and related health · Diet and metabolism studies
АКТУАЛЬНОСТЬ
Врожденный изолированный дефицит АКТГ (ВИДА) — орфанное аутосомно-рецессивное заболевание, обусловленное патогенными вариантами в гене ТВХ19 (MIM 604614), 1q24.2. Фактор транскрипции T-box (TPIT), кодируемый ТВХ19, необходим для терминальной дифференцировки клеток гипофиза, продуцирующих проопиомеланокортин, а также экспрессии гена POMC [1]. Заболевание с одинаковой частотой поражает мальчиков и девочек, в 40–42% отмечаются близкородственные браки [2][3]. В большинстве случаев ВИДА манифестирует в неонатальном периоде тяжелыми гипогликемиями, часто с приступами судорог и обструктивной желтухой [4]. Неспецифичность симптомов и низкая частота заболевания могут привести к поздней диагностике ВИДА [5][6], в связи с чем риск летального исхода в неонатальном периоде достигает 20–25% [3][7].
Представляем двух пациенток с ВИДА с классической манифестацией в неонатальном периоде (желтухой у первой, гипогликемией и апноэ у второй), подтвержденным генетически, причем у одной из них выявлен впервые описанный вариант в гене TBX19.
КЛИНИЧЕСКИЙ СЛУЧАЙ
Пациентка №1
Ребенок от второй физиологической беременности (1 беременность — медицинский аборт), родилась доношенной с массой тела 3750 г, длиной 52 см, малыми аномалиями развития: кранио-фациальные дисморфии и левосторонняя косолапость. Брак близкородственный: родители — двоюродные брат и сестра.
Неонатальный скрининг на врожденный гипотиреоз, врожденную гиперплазию коры надпочечников, галактоземию, фенилкетонурию, муковисцидоз — отрицательный.
В возрасте 1,5 месяца обследована в связи с затяжной желтухой и плохой прибавкой массы тела; отмечалась гипербилирубинемия (общий билирубин — 91 мкмоль/л; норма 8,5–20,5), гиперферментемия (АЛТ — 182,2 Ед/л; норма 5–30, АСТ — 144,5 Ед/л; норма 8–40, ГГТП — 80,8 Ед/л; норма 10–60), гипогликемия без клинических проявлений (глюкоза крови 2,2–2,7 ммоль/л; норма 3,3–5,5). При УЗИ выявлена гепатомегалия, при эластографии — фиброз печени легкой степени. Подтверждено наличие цитомегаловирусной инфекции (повышенный уровень Anti-CMV-IgG, положительная ПЦР на CMV), проведена противовирусная терапия без существенного эффекта.
Пациентка в 7 месяцев экстренно госпитализирована в связи с судорогами. Наблюдались нарушение сознания (сопор), мышечная гипотония, брадикардия (ЧСС 110 уд./мин.) и брадипноэ (ЧД 10 в 1 мин), артериальная гипотония (АД 65/40 мм рт.ст.), иктеричность кожных покровов, гепатоспленомегалия (печень +5,0 см, селезенка +2,0 см из-под реберной дуги), ахоличный стул, темная моча. Отмечалась высокорослость (длина тела — 73 см; SDS 2,03) при дефиците веса (масса тела — 7300 г, SDS ИМТ -2,38). В ходе обследования выявлены тяжелая гипогликемия (глюкоза крови — 0,6 ммоль/л; норма 3,3–5,5), гиперферментемия (АЛТ — 400 Ед/л; норма 5–30, АСТ — 180 Ед/л; норма 8–40), гипербилирубинемия (общий билирубин — 200 мкмоль/л; норма 8,5–20,5, непрямой — 142 мкмоль/л; норма 3,4–12), гипопротеинемия (общий белок — 53 г/л; норма 56–79), анемия (Hb — 97 г/л; норма 114–140, эритроциты — 3,69×10¹²/л; норма 4–5,3), гипокоагуляция (коалиновое время — 97 сек.; норма 50–70, тромбиновое время — 20 сек; норма — 15–18, ПТИ — 63,2%; норма — 78–142, МНО — 1,45; норма — 0,85–1,25, АПТВ — 74 сек.; норма 35–45). Исключено инфицирование Toxoplasma, H. simplex 1, 2, Mycoplasma hominis/pneumoniae, Chlamidia trahomatis/pneumoniae, HBV, HCV, VIH (методом ИФА), ЕВV, CMV, HНV (ПЦР), наследственные болезни обмена: аминоацидопатии, органические ацидемии, дефекты β-окисления жирных кислот (тандемная масс-спектрометрия).
При УЗИ подтверждена гепатоспленомегалия на фоне диффузных изменений паренхимы печени, холестаза.
В процессе дифференциальной диагностики сочетания персистирующей гипогликемии и неинфекционной желтухи выявлены сниженный уровень кортизола (15,9 нмоль/л; норма 138–635) при низком АКТГ (4,99 пг/мл; норма 8,17–46,32). Гиперинсулинизм, нарушение других тропных функций аденогипофиза и гипотиреоз исключены (табл. 1). Это позволило диагностировать ВИДА. Учитывая тяжесть состояния, назначена интенсивная терапия гидрокортизоном парентерально в дозе 75 мг/м²/сут. с последующим постепенным снижением под контролем клинических симптомов и гликемии до поддерживающей 10 мг/м²/сут. в таблетированном виде.
: Таблица 1. Уровни гормонов и глюкозы крови пациентки №1 в 7 месяцевПримечание. ТТГ — тиреотропный гормон, свТ4 — свободный Т4, СТГ — соматотропный гормон, ИФР-1 — инсулиноподобный фактор роста — 1.
При МРТ визуализированы арахноидальная киста левых отделов цистерны продолговатого мозга, диффузная корково-подкорковая атрофия больших полушарий головного мозга.
Для установления причины заболевания проведено секвенирование по Сэнгеру гена TBX19: у пациентки обнаружен ранее описанный [8] патогенный вариант (NM_005149.2.): c.82C>T(p.Q28X) в гомозиготном состоянии, у матери — в гетерозиготном состоянии (рис. 1 б, в).
Рисунок 1. Хроматограммы с фрагментами сиквенса гена TBX19 (позиции с выявленным вариантом указаны стрелкой):а) фрагмент экзона 1 NM_005149.2:c.79_87 (последовательность дикого типа); б) гомозиготная транзиция C>T в положении NM_005149.2:c.82 (пациентка 1); в) гетерозиготная транзиция C>T в положении NM_005149.2:c.82 (мать пациентки 1); г) фрагмент стыка интрон 2/экзон 3 NM_005149.2:c.469-6_473 (последовательность дикого типа); д) гомозиготная транзиция G>A в положении NM_005149.2:c.469-1 (пациентка 2); е) гетерозиготная транзиция G>A в положении NM_005149.2:c.469-1 (отец пациентки 2).
На 5-й день терапии устранена гипогликемия (глюкоза — 4,1 ммоль/л), через 2 месяца разрешилась желтуха, снизилась активность ферментов печени (АЛТ=125 Ед/л, АСТ=93,3 Ед/л, ГГТП=65,1 МЕд/л), через 3 месяца сохранялись ускоренные темпы роста на фоне нормализации массы тела (длина тела — 79 см, SDS +2,57, масса тела — 9200 г, SDS по росту +0,16).
Пациентка №2
Пациентка родилась от пятой беременности, протекавшей на фоне токсикоза и хронической фетоплацентарной недостаточности (старший брат 11 лет — здоров), вторых родов на 38-й неделе с массой тела 3340 г, длиной 52 см. Брак неродственный. В первые сутки отмечался эпизод апноэ, на вторые двукратно выявлена гипогликемия (глюкоза крови — 1,1 и 2,4 ммоль/л). В 2,5 месяца госпитализирована в неврологическое отделение в связи с повторным эпизодом апноэ и гипогликемическими судорогами (гликемия — 2,0 ммоль/л). Проведена КТ головного мозга, выявлены признаки смешанной гидроцефалии, субатрофии вещества головного мозга. Причина гипогликемии не установлена. Рекомендована вальпроевая кислота. В 3,5 месяца при плановом обследовании выявлены гипокортизолемия (кортизол — 27,6 нмоль/л; норма 138–635), умеренные гипертиреотропин- и гипотироксинемия (ТТГ — 8,9 мМЕ/л; норма 0,62–8,0, свТ4 — 9,05 пмоль/л; норма 11,5–20,4). В 4 месяца отмечалась высокорослость (длина тела — 68 см; SDS 3,1) при дефиците веса (масса тела — 6400 г, SDS ИМТ -2,6). В 7,5 месяцев госпитализирована в неврологическое отделение в связи с приступом тонико-клонических судорог на фоне поствакцинальной лихорадки. В ходе обследования неоднократно отмечалась гипогликемия (минимальная глюкоза крови — 0,59 ммоль/л). Диагностированы инфекционно-аллергический энцефалит (тяжелая форма), симптоматическая эпилепсия. В возрасте 1 года 9 месяцев повторно выявлена гипокортизолемия (кортизол — 24,4 нмоль/л) при низконормальном АКТГ 3,68 пг/мл (норма 0–30), диагностирован центральный гипокортицизм, начата терапия гидрокортизоном 15 мг/м²/сут. Подтвержден субклинический гипотиреоз (ТТГ — 16,3 мМЕ/л, свТ4 — 14,3 пмоль/л), назначен левотироксин — 12,5 мкг/сут.
В 2 года 1 месяц госпитализирована для контроля терапии и уточнения диагноза с жалобами на задержку психомоторного развития: начала ходить после года, не произносит слов. На фоне терапии гидрокортизоном в прежней дозе сохраняется высокорослость (рост — 98 см, SDS 3,8) и высокая скорость роста (19,1 см/год; SDS 3,9), масса тела нормализовалась (вес — 17 кг, SDS ИМТ 0,9); достигнута нормогликемия (глюкоза — 3,66–4,01 ммоль/л). Несмотря на нерегулярный прием левотироксина в минимальной дозе субклинический гипотиреоз не прогрессирует (ТТГ — 10,6 мМЕ/л; норма 0,64–5,76, свТ4 — 13,2 пмоль/л; норма 11,5–20,4). Проведено секвенирование по Сэнгеру гена TBX19, у пациентки обнаружен ранее не описанный в HGMD патогенный вариант (NM_005149.2.): c.469-1G>A в гомозиготном состоянии, у обоих родителей — в гетерозиготном состоянии (рис. 1 д, е).
ОБСУЖДЕНИЕ
ВИДА вследствие патогенных вариантов в гене ТВХ19 крайне редкое заболевание, данные по заболеваемости и распространенности ограничены. В мировой литературе в основном освещаются единичные клинические случаи, которых насчитывается не более 80. Впервые доказательства связи ВИДА с дефектами в гене ТВХ19 были представлены в 2001 г. Lamolet с соавт., показавшими коэкспрессию генов Tpit и Pomc в клеточных линиях гипофиза мыши, и выявившими мутации в гене TPIT (TBX19) у 2 пациентов с вторичной надпочечниковой недостаточностью [9]. Abali Z.Y. и соавт. (2019 г.) обобщили данные о 66 ранее описанных пациентах, среди которых у большинства симптомы развивались в неонатальном периоде и заболевание диагностировалось до 2 лет [2][3][8].
Первые симптомы заболевания неспецифичны, что значительно затрудняет диагностику ВИДА [8]. Наиболее частыми симптомами считают гипогликемию (100%), в том числе симптоматическую (судороги, 53%), холестатическаую желтуху (62%), а также гипотермию или лихорадку, шок [3]. По данным ряда авторов, один из первых симптомов — приступ апноэ на 1–11-й дни жизни [1][10][11]. У некоторых пациентов развивается особая форма гепатита — неонатальный неинфекционный гепатит, связанный с гипокортизолемией; диагноз подтверждается при биопсии почти в каждом втором случае сочетания холестатической желтухи и гепатомегалии [8][11].
И в наших случаях диагноз установлен только на 8-м и 22-м месяцах жизни, несмотря на появление клинически значимых симптомов в периоде новорожденности у обеих пациенток, хотя гипогликемические судороги у пациентки №1 развились лишь к 7 месяцам, а у пациентки №2 отсутствовали признаки холестаза. Эпизод апноэ в первые сутки жизни у пациентки №2 следует расценить как результат гипогликемии. У пациентки №1 основными проявлениями заболевания были холестатическая желтуха (ахоличный стул, темная моча, гипербилирубинемия), признаки цитолиза гепатоцитов (гиперферментемия), нарушение белковосинтетической функции печени (гипоальбуминемия, гипокоагуляция), гепатомегалия. И хотя биопсию печени не проводили, данные изменения и отсутствие эффекта от противовирусной терапии могут свидетельствовать о развитии неинфекционного холестатического гепатита. Связь поражения печени с гипокортизолемией подтверждают улучшение и постепенная нормализация клинико-лабораторных изменений на фоне терапии гидрокортизоном. Учитывая вышесказанное, целесообразно при наличии гипогликемии ± холестаза у новорожденного или младенца исключить гипокортицизм.
Данные о росте пациентов с ВИДА вследствие патогенных вариантов в гене ТВХ19 противоречивы. Некоторые авторы описывают высокорослость на фоне нормального или сниженного ИФР-1 с нормализацией темпов роста на фоне заместительной терапии глюкокортикоидами [2][4][6]. Другие исследователи не выявили изменений антропометрических показателей у пациентов [12][13]. У обеих описываемых пациенток отмечалась высокорослость на фоне дефицита веса: умеренной степени к 7 месяцам (пациентка №1) и тяжелой в возрасте 4 месяцев (пациентка №2). Нормальный уровень СТГ и низконормальный — ИФР1 у пациентки №1 исключают связь высокорослости с нарушением в системе ГР-ИФР1. В отличие от литературных данных, высокие темпы роста на фоне терапии в течение 3–4 месяцев сохранялись при нормализации массы тела. Ввиду невозможности исследования уровня ИФР1-связывающего белка 5 подтвердить или опровергнуть гипотезу о связи высокорослости с отсутствием ингибирования этого белка при дефиците глюкокортикоидов, обусловленном ВИДА, не удалось [2].
Ряд авторов сообщали о различных дисморфологических признаках у пациентов с ВИДА, таких, как порок Арнольда-Киари 1-го типа и черепно-лицевые малые аномалии [3][4][6]. У пациентки №1 отмечались тригоноцефалия, скошенный затылок, узкие глазные щели, эпикант, антимонголоидный разрез глаз, широкая плоская переносица, микростомия, готическое небо, низко посаженные уши, левосторонняя косолапость. У пациентки №2 подобные аномалии не наблюдались. Установить связь фенотипа с патогенным вариантом гена ТВХ19 не представляется возможным. У обеих пациенток при визуализации выявлена диффузная атрофия/субатрофия вещества головного мозга, у пациентки №1 дополнительно — арахноидальная киста, у пациентки №2 — гидроцефалия. Подобных изменений ЦНС в литературе не описано, хотя в некоторых случаях отмечали умственную отсталость. Исключить связь субатрофии/атрофии вещества головного мозга у наших пациенток с тяжелыми гипогликемиями не представляется возможным.
Для ВИДА характерны резко сниженный уровень кортизола при сниженном [2][3][4][7][8][10][12] или нормальном [1][6] АКТГ. Следует отметить, что оценка уровня АКТГ зависит от чувствительности тест-систем. У наших пациенток отмечались аналогичные результаты: низкие уровни кортизола при сниженном или низконормальном АКТГ.
Среди пациентов с ВИДА описано несколько случаев идиопатического субклинического гипотиреоза [5][6][7][9]. В последующем уровни ТТГ нормализовались, в том числе спонтанно или на фоне лечения левотироксином в минимальных дозах. У пациентки №2 также была выявлена гипертиреотропинемия (в 3,5 месяца), сохраняющаяся в 2 года 1 месяц на фоне нерегулярного приема левотироксина 12,5 мкг/сут.
По данным на 2021 г., в HGMD (http://www.hgmd.cf.ac.uk) включены 29 патогенных вариантов в гене TBX19: ряд миссенс-мутаций, большие и малые делеции, четыре мутации в пределах сайта сплайсинга и новая синонимичная мутация NM_005149.3:c.288G>A (p.T96=) [1].
У пациентки №1 обнаружен патогенный вариант c.82C>T(p.Q28X) в гомозиготном состоянии, ранее выявленный Vallette-Kasic S и соавт., 2005 [8], что позволило подтвердить диагноз «ВИДА». Авторы описали сходные проявления у родных брата и сестры с данной мутацией: гипогликемию с судорогами, холестатическую желтуху, однако гепатит подтвержден не был и не отмечены какие-либо малые аномалии развития.
У пациентки №2 выявлен ранее не описанный вариант в гене ТВХ19 c.469-1G>A в гомозиготном состоянии. Хотя в настоящее время данный вариант отсутствует в референтных популяционных базах данных (https://gnomad.broadinstitute.org/), он может быть расценен как патогенный и каузальный, так как относится к влияющим на сплайсинг и присутствует в гомозиготном состоянии у пациента с классической клинико-лабораторной картиной ВИДА.
ЗАКЛЮЧЕНИЕ
Представленные нами клинические случаи подчеркивают, что при наличии персистирующей гипогликемии, особенно в сочетании с холестазом, у новорожденного или младенца необходимо исключить гипокортицизм.
Выявление врожденного изолированного дефицита АКТГ (двукратно низкий уровень кортизола в сочетании с неповышенным АКТГ при отсутствии нарушения других функций аденогипофиза) требует молекулярно-генетического исследования.
В одном из представленных в настоящей публикации случаев выявлен ранее не описанный вариант в гене ТВХ19 (NM_005149.2.): c.469-1G>A.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Источники финансирования. Молекулярно-генетическое исследование выполнено при частичном содействии Фонда поддержки и развития филантропии «КАФ».
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Согласие пациента. Пациенты добровольно подписали информированное согласие на публикацию персональной медицинской информации в обезличенной форме в журнале «Проблемы эндокринологии».
Благодарности. Выражаем благодарность Иванову Дмитрию Владимировичу, врачу педиатру, за своевременный диагностический поиск и оказание квалифицированной экстренной врачебной помощи пациентке №1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Maudhoo Ashwini Maharaj Avinaash Buonocore Federica Martos-Moreno Gabriel Angel Argente Jesús Achermann John C Chan Li F Metherell Lou A Missplicing due to a synonymous, T 96= exonic substitution in the T-box transcription factor TBX 19 resulting in isolated ACTH deficiency Endocrinology, Diabetes & Metabolism Case Reports 202110202110.1530/edm-21-0128 PMC 849572334564059 · doi ↗ · pubmed ↗
- 2Abali Zehra Yavas Yesil Gozde Kirkgoz Tarik Kaygusuz Sare Betul Eltan Mehmet Turan Serap Bereket Abdullah Guran Tulay Evaluation of growth and puberty in a child with a novel TBX 19 gene mutation and review of the literature Hormones 20190222923618210.1007/s 42000-019-00096-730747411 · doi ↗ · pubmed ↗
- 3Couture C.Saveanu A.Barlier A.Carel J. C.Fassnacht M.Flück C. E.Houang M.Maes M.Phan-Hug F.Enjalbert A.Drouin J.Brue T.Vallette S.Phenotypic Homogeneity and Genotypic Variability in a Large Series of Congenital Isolated ACTH-Deficiency Patients with TPIT Gene Mutations The Journal of Clinical Endocrinology & Metabolism 201112 E 486E 49597310.1210/jc.2011-165922170728 · doi ↗ · pubmed ↗
- 4Kardelen Al Aslı Derya PoyrazoğluŞükran Aslanger Ayça Yeşil Gözde Ceylaner Serdar BaşFirdevs Darendeliler Feyza A Rare Cause of Adrenal Insufficiency – Isolated ACTH Deficiency Due to TBX 19 Mutation: Long-Term Follow-Up of Two Cases and Review of the Literature Hormone Research in Paediatrics 20200439540392610.1159/00050674032344415 · doi ↗ · pubmed ↗
- 5Vieira Inês Henriques Mourinho Bala Nádia Ramos Fabiana Dinis Isabel Cardoso Rita Caetano Joana Serra Rodrigues Dírcea Paiva Isabel Mirante Alice A serious and unusual presentation of congenital isolated ACTH deficiency due to TBX 19 mutation, beyond the neonatal period Endocrinology, Diabetes & Metabolism Case Reports 202209202210.1530/edm-22-0277 PMC 951365536070412 · doi ↗ · pubmed ↗
- 6Weijing Kong Liping Zou Tiantian Zhang Pei Zhang Yan Meng A Case of Congenital Isolated Adrenocorticotropic Hormone Deficiency Caused by Two Novel Mutations in the TBX 19 Gene Frontiers in Endocrinology 2019041010.3389/fendo.2019.00251 PMC 648225831057487 · doi ↗ · pubmed ↗
- 7Peng Cheng Sun Guoyu Tang Zezhong Hou Xinlin Congenital Isolated ACTH Deficiency Caused by TBX 19 Gene Mutation: A Family Report Frontiers in Pediatrics 202001710.3389/fped.2019.00546 PMC 696741631998673 · doi ↗ · pubmed ↗
- 8Vallette-Kasic Sophie Brue Thierry Pulichino Anne-Marie Gueydan Magali Barlier Anne David Michel Nicolino Marc Malpuech Georges Déchelotte Pierre Deal Cheri Van Vliet Guy De Vroede Monique Riepe Felix G.Partsch Carl-Joachim Sippell Wolfgang G.Berberoglu Merih Atasay Begümde Zegher Francis Beckers Dominique Kyllo Jennifer Donohoue Patricia Fassnacht Martin Hahner Stefanie Allolio Bruno Noordam C.Dunkel Leo Hero Matti Pigeon B.Weill Jacques Yigit Sevket Brauner Raja Heinrich Juan Jorge Cummings Elizabeth Riddell Christie Enjalbert Alain Drouin Jacques Congenital · doi ↗ · pubmed ↗