Chromosome 1p31.1 Deletion: A Case With Developmental Delay, Hypotonia, Cryptorchidism, Abnormal Oral Frenulum, and Feet Deformity
Tatiana Mikhailova, Ria Garg

TL;DR
This paper reports a rare chromosomal deletion case with unique features like abnormal oral frenulum and feet deformity.
Contribution
The study adds a new case of 1p31.1 deletion with a unique combination of clinical features, including thickened oral frenulum.
Findings
A 14.385 Mb deletion in 1p31.1 encompassing 41 genes was identified.
The case exhibited microcephaly, hypotonia, developmental delay, and flat feet.
Congenital thickening of the lingual and labial frenulum was observed, a feature not previously associated with this deletion.
Abstract
Deletions within the chromosomal locus 1p31.1 are rare, with only a limited number of documented cases. The typical clinical presentation includes intellectual disability, failure to thrive, and craniofacial abnormalities. Some cases may also present with cardiac, gastrointestinal, and genitourinary malformations. Variability in deletion size contributes to a broad spectrum of clinical phenotypes, and a comprehensive understanding of the syndrome's manifestations is still evolving. This case study aims to provide additional insights into 1p31.1 microdeletion syndrome, enhancing knowledge of its genetic and phenotypic characteristics to improve recognition by clinicians. Here, we report a case featuring a 14.385 Mb deletion isolated to the 1p31.1 region, encompassing 41 genes. The deletion manifested with microcephaly, distinctive facial morphology, hypotonia, developmental delay,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
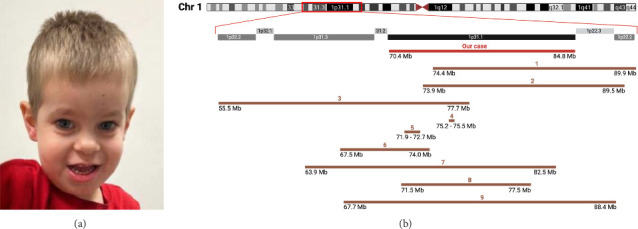 Figure 1
Figure 1- —National Institutes of Health
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer and Skin Lesions · Genomic variations and chromosomal abnormalities · Hedgehog Signaling Pathway Studies
1. Introduction
The chromosome 1p31.1 deletion is among the less described microdeletion syndromes, typically presenting with developmental delay, intellectual disability, craniofacial malformations, and other systemic abnormalities [1–3]. In 1979, Bene et al. reported the case of a 14 year-old girl with severe psychomotor delay, short stature, and dysmorphic features linked to a partial deletion of the short arm of Chromosome 1 [4]. Subsequent case reports have documented additional individuals with similar deletions, characterized by developmental delay and various congenital malformations [5–10]. Advances in genomic technology have enabled more precise localization and annotation of the affected genomic regions. Despite the phenotypic similarities across cases, understanding the genotype–phenotype relationship has been challenging due to variability in deletion size and breakpoints, which leads to involvement of different genes in each case [1, 3, 11–13].
This case study reports the first documented occurrence of a de novo deletion spanning 14.385 Mb, encompassing the genomic coordinates 70,438,241–84,822,837 on the p-arm of Chromosome 1. Alongside the established syndrome-associated traits such as microcephaly, hypotonia, and delays in psychomotor and speech development, we describe uncommon presentations of early postnatal bilateral cryptorchidism and thickening of the lingual and labial frenulum. These manifestations, while not emphasized in prior case studies, could be notable features of 1p31.1 deletion. We provide a comprehensive clinical overview of the case, contrasting the observed phenotypic profile with those documented in previous case studies with deletion coordinates mapped to the 1p31.1 locus.
2. Case Description
The patient described in this report is a 2-year-old male, delivered at full term to a 24-year-old mother (G2P0 > 1) and a 25-year-old nonconsanguineous father. The pregnancy was complicated by maternal hypertension but was otherwise unremarkable, with no infections, diabetes, medication use, or exposure to tobacco, alcohol, or drugs. Fetal ultrasounds showed normal development and movements. Delivery was complicated by a cesarean section due to failed induction. Birth measurements were as follows: length 50.8 cm (23rd percentile), weight 2667 g (7th percentile), and head circumference 32 cm (5th percentile), with Apgar scores of 8 and 9 at 1 and 5 min, respectively. No abnormalities were noted at birth.
On the third day after birth, the patient presented to the emergency department with breathing difficulties following milk aspiration. Evaluation revealed hypothermia, hypoglycemia, and respiratory distress, necessitating a 5-day stay in the neonatal intensive care unit (NICU). In the NICU, ankyloglossia and a tight upper labial frenulum were noted, prompting lingual and upper labial frenulectomies. Partial recurrence of the banding was observed at a 3-month follow-up, though no repeat procedure was recommended due to lack of functional impairment.
At the 2-month pediatric evaluation, the patient's development was reported as normal, with no concerns identified. At 4 months, however, hypotonia and motor developmental delays were observed, prompting a referral to the child neurology clinic. Evaluation by the neurologist revealed generalized hypotonia, difficulty with head and chest lifting, lack of coordination, poor weight gain, and borderline microcephaly (−2 SD). His measurements were as follows: height 64.3 cm (47th percentile), weight 5.05 kg (< 1st percentile), and head circumference 38.8 cm (< 1st percentile). Physical examination showed dysmorphic features, including a long face with a narrow and tall forehead, closely spaced eyes, a high-arched narrow palate, a broad nasal tip, and micrognathia. Given these findings, a genetic etiology was suspected, and chromosomal microarray testing was ordered.
At 10 months, the patient was evaluated by pediatric urology for bilateral undescended testicles, requiring bilateral orchidopexy. An ophthalmology assessment at that time revealed hyperopia. At 19 months, an ear, nose, and throat (ENT) examination found malalignment of mandibular incisors, likely due to the initial ankyloglossia, contributing to dental malocclusion. A musculoskeletal evaluation at age 2 by the orthopedic surgery department identified bilateral flat feet and partial cutaneous syndactyly between the second and third toes, characterized by the second toe overlapping the third. The developmental history indicated delays in motor milestones, with head control achieved at 5 months, independent sitting at 7 months, crawling at 9 months, and walking at 18 months.
Genetic screening results were delayed due to a transfer of care between hospitals. At 2 years, chromosomal microarray analysis revealed a pathogenic 14.385 Mb deletion on chromosome 1p31.1 (70,438,241–84,822,837), involving 41 protein-coding genes. On follow-up evaluation at the pediatric genetics department, previously documented dysmorphic features and an open-mouth posture were noted (Figure 1(a)). Language delay was noted, with the patient speaking his first words at 12 months. During evaluation, he had a limited vocabulary and was unable to form short sentences. He also displayed episodic impulsivity and an abnormal gait. Growth parameters showed head circumference below the 5th percentile, with height and weight within normal ranges.
Given the limited knowledge of the syndrome, management recommendations focused on addressing symptomatic concerns through a multidisciplinary approach. A renal ultrasound and cardiological evaluation, including an EKG and echocardiogram, were performed, all yielding unremarkable findings. At a neurological evaluation at 2 years 11 months, the patient's height and weight were in the 30th and 23rd percentiles, respectively, with a head circumference below the 2nd percentile. He was described as friendly, enjoyed interacting with others, showed no repetitive behaviors or stereotyped movements, maintained good eye contact, and exhibited no sensory issues. However, delayed speech development was noted; his vocabulary consisted of 20–30 words, and he primarily communicated in 1–2 word phrases, with echolalia also observed. Physical examination revealed diffusely low muscle tone and hypermobile joints. While he exhibited no difficulties with walking or running, he demonstrated poor balance and coordination. To address these challenges, the patient was enrolled in early intervention programs, including speech, physical, and occupational therapy.
3. Discussion
The individual presented in this case report has a notable deletion within the chromosome 1p31.1 locus, encompassing 41 genes, with the distal breakpoint located within the LRRC7 coding region (Supporting Table 1). Hallmark features of 1p31.1 deletion include intellectual disability, delayed psychomotor development, speech articulation difficulties, hypotonia, growth delay, and dysmorphic facial features such as a prominent forehead, microcephaly, micrognathia, broad nasal tip, and epicanthal folds (Figure 1(a) and Table 1). Similar clinical characteristics, including genitourinary anomalies and foot abnormalities, have been observed in cases from the DECIPHER database (Table 2). Although inherited cases of the deletion have been documented [12, 16], the majority arise from de novo mutations [2, 4], sometimes due to genetic translocation events in healthy parents [13]. In our case, parental testing for the deletion yielded normal results, confirming its de novo origin. Testing for potential balanced rearrangements in parental samples was not conducted.
The low incidence of 1p31.1 deletion, variability in the deleted genomic intervals, and potential pleiotropic effects obscure understanding of how the absence of specific genes contributes to the observed phenotype. However, it is apparent that the severity of symptoms exhibits a dose-dependent effect, with more extensive deletions associated with more pronounced clinical presentations (Table 1). Larger deletions of approximately 20 Mb within 1p32.2–1p22.1 loci resulted in severe craniofacial abnormalities and congenital malformations that impact fetal viability (Figure 1(b) and Table 1) [11–13]. Intermediate deletions within the 15 Mb range were not discernible during prenatal and early postnatal evaluations. Affected individuals manifested symptoms around 4–6 months of age, characterized by psychomotor delay evident as an inability to support the head. This pattern was observed in our case, as well as in a previously described report involving a 15.5 Mb deletion spanning from 1p31.1 to 1p22.2 [14]. The deletion of a single gene or a short noncoding sequence may result in phenotypes insufficiently apparent to suggest a genetic etiology during early childhood [3, 16]. For example, an individual with a single gene deletion did not undergo evaluation for genetic defects until concerns regarding learning impairment emerged at 7 years of age, even when impulsivity, hyperactivity, and speech delays were noted at 13 months [3]. A smaller microdeletion of 0.27 Mb lacking protein-coding genes did not result in any abnormalities except thickened labial frenulum and dental misalignment [16]. Frenulum abnormalities were also observed in our case, requiring surgical intervention, as tight frenula in newborns can impair tongue mobility, interfere with breastfeeding, and contribute to early nutritional deficiencies [17].
Cognitive delay and neurological manifestations, including speech and psychomotor disturbances, as well as attention and memory deficits, are the most common manifestations of 1p31.1 deletion (Table 1). Rivera-Pedroza et al. provided detailed insight into a case featuring a 18.6 Mb 1p31.1 through p31.3 deletion, which presented severe physical and developmental challenges [12]. In addition, Biswal et al. described a case involving an 11-year-old male who had a 5.99 Mb deletion at 1p31.1, which included 18 genes that are also absent in our case. The individual described by the authors demonstrated moderate intellectual disability, inattention, hyperactivity, and severe language impairment, with the emergence of first words only at age 5 and the ability to form short sentences achieved only at age 11 [1]. These difficulties mirror those observed in our case and other previous reports (Table 1). The study by Callier et al. provides insight into the long-term developmental trajectory of individuals with 1p31.1 deletion, describing a 20-year-old male with a 15.6 Mb deletion who exhibited moderate intellectual disability and remained unable to write, read, or count [15].
The observed neurological manifestations are likely attributable to a multifaceted interplay among numerous genes. The neuronal growth regulator (NEGR1), located at 71,861,626–72,748,222 on Chromosome 1 (Supporting Table 1), is frequently observed within the deleted intervals and has been previously described in the context of 1p31.1 deletion [3]. All but one of the documented cases in our study exhibit a NEGR1 deletion, consistently presenting with psychomotor and developmental delays (Figure 1(b) and Table 1). Genovese et al. reported a case of two siblings with a deletion in the 71,868,625–72,748,533 region, affecting only the NEGR1 gene, presenting with developmental and language delays, psychomotor retardation, and hypotonia [3]. The mechanism by which NEGR1 deletion could contribute to these phenotypes is not fully understood due to limited human evidence. Mouse models have shown that NEGR1 deficiency disrupts neurite outgrowth during neuritogenesis and alters the excitatory/inhibitory neuronal balance, resulting in social behavior impairments [18]. The NEGR1 protein has also been shown to interact with fibroblast growth factor receptor 2 (FGFR2), influencing cortical development via the ERK and AKT signaling pathways [19].
The distal breakpoint of the deletion we report falls within the LRRC7 gene, resulting in the loss of 19 out of 25 exons. LRRC7 encodes densin-180, a protein involved in maintaining postsynaptic neuronal signaling through interactions with signaling proteins, such as the α-subunit of Ca2+/calmodulin-dependent protein kinase II (αCaMKII) [20]. Truncations of the LRRC7 gene result in a loss of protein function, as its C-terminal residues are essential for binding to αCaMKII, which modulates phosphorylation of synaptic proteins during synaptic plasticity. Individuals with loss-of-function variants in LRRC7 consistently exhibit a phenotype characterized by intellectual disability, attention deficits, and global neurodevelopmental delay, including delays in motor and speech development [20].
Although cryptorchidism has rarely been reported as a characteristic of 1p31.1 deletion syndrome, it has been previously described by Callier et al. [15] and documented in the DECIPHER database in individuals with a similar deletion region (Tables 1 and 2). While the etiology of cryptorchidism remains unclear, the 1p31.1 deletion could plausibly disrupt hypothalamic–pituitary signaling, supported by a documented case of congenital hypopituitarism associated with 1p31.1 deletion [9]. Considering the potential consequences of failed testicular descent, such as an increased risk of testicular neoplasms and torsion, it is important to recognize cryptorchidism as a possible component of the syndrome and ensure timely management [21, 22].
The absence of a standardized management protocol for 1p31.1 deletion syndrome necessitates individualized treatment plans. Comprehensive developmental and neuropsychological assessments are essential, including speech therapy and evaluations of neurological, visual, and auditory systems. Palate defects, genitourinary abnormalities, abnormal muscle tone, growth restriction, and low-set ears occur in over 50% of cases and should be routinely assessed during clinical follow-up. Less common but clinically significant findings, such as cardiac anomalies, neuroimaging abnormalities, musculoskeletal issues (e.g., scoliosis, joint laxity, and toe deformities), seizures, and dental anomalies, should be evaluated and monitored based on clinical judgment. Among facial dysmorphic features, low-set ears, micrognathia, and epicanthal folds are present in over half of affected individuals, supporting their use as key features in diagnostic evaluation and dysmorphology assessments (Table 3). The variability of other clinical manifestations further underscores the need for personalized management strategies (Figure 1(a) and Table 1).
In conclusion, we report a case of a 14.385 Mb deletion in the 1p31.1 region which, in addition to developmental challenges including hypotonia and language delays, also presented with cryptorchidism and thickened frenula. These findings expand the phenotypic spectrum of 1p31.1 deletion syndrome by adding features not previously reported. We aim to contribute to the knowledge of this rare deletion to improve the diagnosis and management of affected patients, emphasizing their need for multidisciplinary care.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Biswal S. Parida P. Dubbudu A. Sharawat I. K. Panda P. K. Chromosome 1p 31.1 Deletion Syndrome: Limited Expression Annals of Indian Academy of Neurology 2021241788010.4103/aian.AIAN_258_2033911383 PMC 8061518 · doi ↗ · pubmed ↗
- 2Tassano E. Gamucci A. Celle M. E. Ronchetto P. Cuoco C. Gimelli G. Clinical and Molecular Cytogenetic Characterization of a De Novo Interstitial 1p 31.1p 31.3 Deletion in a Boy With Moderate Intellectual Disability and Severe Language Impairment Cytogenetic and Genome Research 20151461394310.1159/0004313912-s 2.0-8494193701726112959 · doi ↗ · pubmed ↗
- 3Genovese A. Cox D. Butler M. Genovese A. Partial Deletion of Chromosome 1p 31.1 Including Only the Neuronal Growth Regulator 1 Gene in Two Siblings Journal of Pediatric Genetics 2015040102302810.1055/s-0035-1554977 PMC 490641427617112 · doi ↗ · pubmed ↗
- 4Bene M. Duca-Marinescu A. Ioan D. Maximilian C. De Novo Interstitial Deletion del(1)(p 21p 32) Journal of Medical Genetics 197916432332710.1136/jmg.16.4.323490590 PMC 1012682 · doi ↗ · pubmed ↗
- 5Lai M. M. Robards M. F. Berry A. C. Fear C. N. Hart C. Two Cases of Interstitial Deletion 1p Journal of Medical Genetics 199128212813010.1136/jmg.28.2.1282002484 PMC 1016783 · doi ↗ · pubmed ↗
- 6Petersen M. B. Warburg M. Interstitial Deletion 1p in a 30-Year-Old Woman Journal of Medical Genetics 198724422923110.1136/jmg.24.4.2292953897 PMC 1050002 · doi ↗ · pubmed ↗
- 7Stockton D. W. Ross H. L. Bacino C. A. Altman C. A. Shaffer L. G. Lupski J. R. Severe Clinical Phenotype Due to an Interstitial Deletion of the Short Arm of Chromosome 1: A Brief Review American Journal of Medical Genetics 199771218919310.1002/(SICI)1096-8628(19970808)71:2<189::AID-AJMG 13>3.0.CO;2-A 9217220 · doi ↗ · pubmed ↗
- 8Mattia F. R. Wardinsky T. D. Tuttle D. J. Grix A. Smith K. A. Walling P. Interstitial Deletion of the Short Arm of Chromosome 1 (46XY, del(1)(p 13p 22.3)) American Journal of Medical Genetics 199244555155410.1002/ajmg.13204405032-s 2.0-00264852121481806 · doi ↗ · pubmed ↗