Muhenrins A–C, pimarane-type diterpenoids from Munronia henryi
Wan-Bai Su, Xiao-Meng Hou, Ling Zhang, Li Yan, Zhi-Yang Tang, Yang Yu, Jin-Song Liu, Yun-Peng Sun, Guo-Kai Wang

TL;DR
Researchers discovered three new diterpenoids from a plant called Munronia henryi and studied their structures and potential anti-inflammatory effects.
Contribution
The discovery of three new pimarane-type diterpenoids and their structural elucidation with biosynthetic insights.
Findings
Muhenrin A has a unique 6,7-seco pimarane skeleton with proposed biosynthetic pathways.
Compounds 1–4 showed weak inhibition of NO production in LPS-induced RAW 264.7 cells.
Structures were confirmed using spectroscopic methods and X-ray diffraction.
Abstract
Three undescribed pimarane-type diterpenoids, muhenrins A–C (1–3), along with a known analogue were isolated from the petroleum ether extract of Munronia henryi (Meliaceae). Their structures, including absolute configurations, were elucidated by means of various spectroscopic methods (IR, UV, HR-ESI-MS, NMR), single-crystal X-ray diffraction, ECD and NMR calculations. Muhenrin A (1) features a unique 6,7-seco pimarane skeleton, and its putative biosynthetic pathways have been proposed. Compounds 1–4 showed weak inhibitory activity against NO production in LPS-induced RAW 264.7 cells, with inhibition rates ranging from 13.73 – 32.35% at 50 μM concentrations. Three pimarane-type diterpenoids, muhenrins A–C (1–3), along with a known analogue were isolated from Munronia henryi (Meliaceae).
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
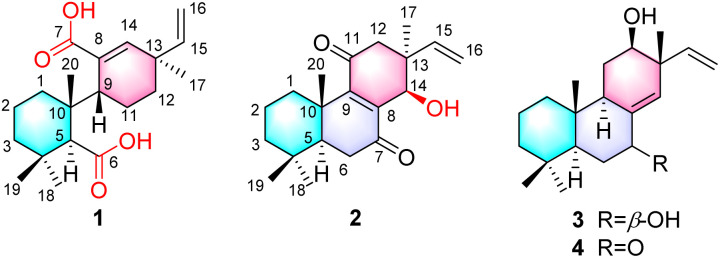 Fig. 1
Fig. 1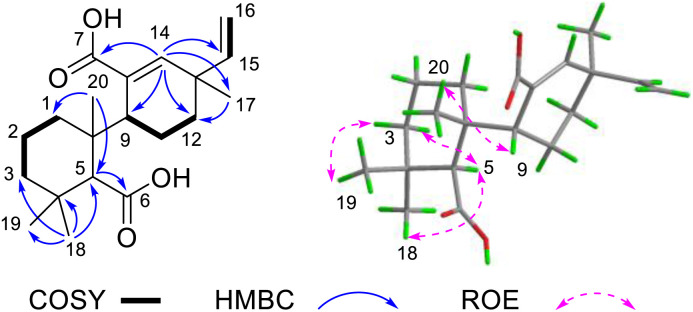 Fig. 2
Fig. 2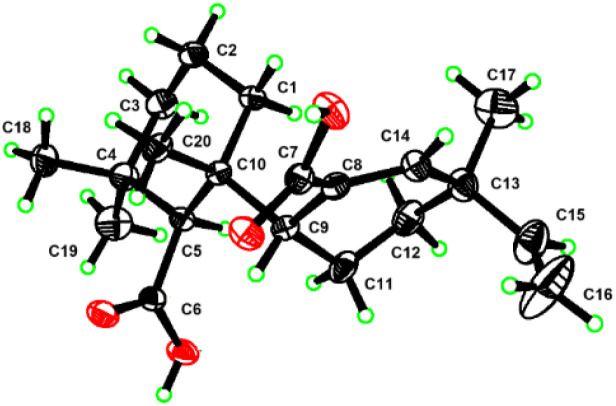 Fig. 3
Fig. 3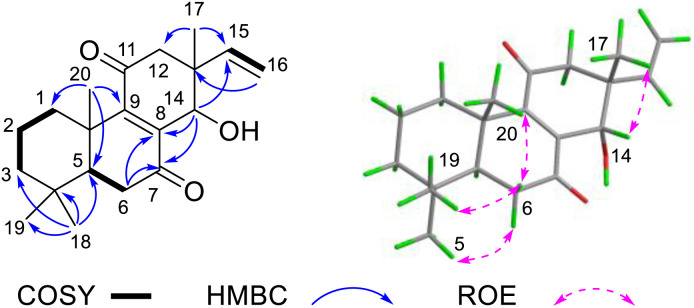 Fig. 4
Fig. 4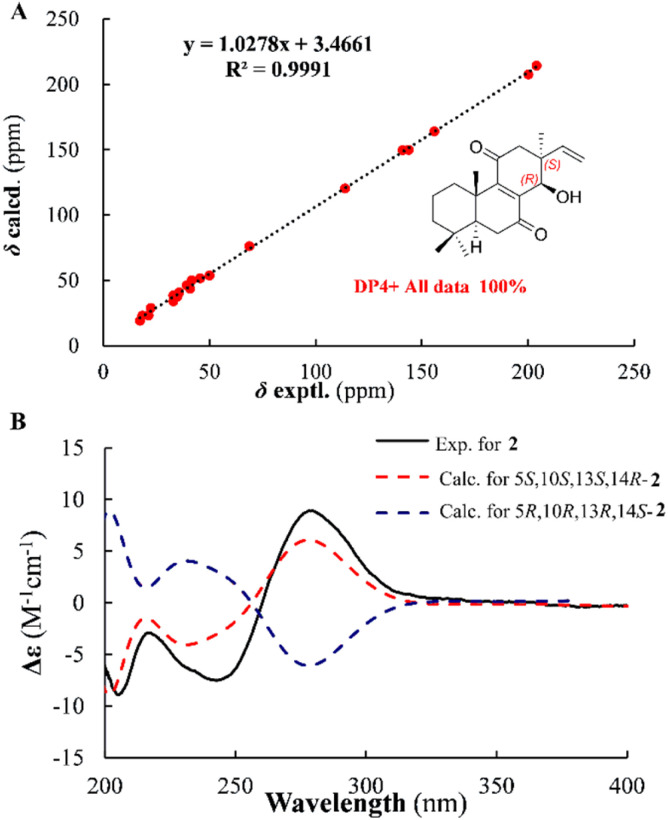 Fig. 5
Fig. 5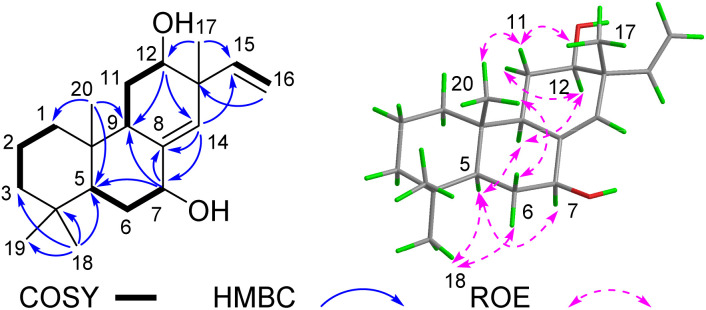 Fig. 6
Fig. 6| No | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
|
|
|
|
|
|
| |
| 1α | 36.5 | 1.49, m | 35.0 | 0.96, dd (13.2, 3.7) | 39.1 | 1.00, m |
| 1β | 1.37, m | 2.69, br d (13.2) | 1.66, m | |||
| 2α | 19.8 | 1.50, m | 18.5 | 1.52, m | 19.0 | |
| 2β | 1.62, m | 1.70, qt (13.7, 3.5) | 1.48, m | |||
| 3α | 42.0 | 1.19, m | 40.9 | 1.20, dd (13.6, 3.9) | 41.9 | 1.17, td (13.3,4.5) |
| 3β | 1.53, m | 1.46, br d (13.4) | 1.45, m | |||
| 4 | 34.5 | 33.1 | 33.3 | |||
| 5 | 58.6 | 2.55, s | 50.1 | 1.59, dd (15.0, 3.2) | 52.0 | 3.87, t (8.0) |
| 6α | 177.1 | 35.7 | 2.56, dd (17.8, 3.2) | 32.6 | 2.00, m | |
| 6β | 2.44, dd (17.8, 15.0) | 1.29, m | ||||
| 7 | 174.9 | 204.1 | 72.5 | 3.99, br s | ||
| 8 | 135.3 | 141.1 | 139.4 | |||
| 9 | 41.6 | 2.78, br s | 156.0 | 50.3 | 1.93, m | |
| 10 | 42.7 | 39.4 | 38.4 | |||
| 11α | 21.5 | 1.60, m | 200.3 | 26.3 | 1.80, m | |
| 11β | 2.06, br d (14.7) | 1.53, m | ||||
| 12α | 33.8 | 1.54, m | 45.7 | 2.53, d (17.0) | 73.2 | 3.57, dd (12.3, 3.9) |
| 12β | 1.74, td (13.0, 3.0) | 2.65, d (17.0) | ||||
| 13 | 38.9 | 41.7 | 42.7 | |||
| 14 | 144.7 | 6.33, s | 68.9 | 4.55, d (2.6) | 124.9 | |
| 15 | 147.1 | 5.73, dd (17.4, 10.6) | 143.9 | 5.75, dd (17.6, 10.9) | 146.2 | 5.80, dd (17.4, 10.8) |
| 16a | 112.7 | 4.82, d (17.6) | 113.9 | 4.93, d (17.6) | 114.1 | 5.14, d (17.4) |
| 16b | 4.96, d (10.6) | 5.04, d (10.9) | 5.15, d (10.8) | |||
| 17 | 27.2 | 1.14, s | 22.4 | 1.16, s | 17.5 | 1.08, s |
| 18 | 34.7 | 1.03, s | 33.0 | 0.89, s | 33.8 | 0.91, s |
| 19 | 24.5 | 1.16, s | 21.4 | 0.93, s | 22.2 | 0.87, s |
| 20 | 22.5 | 1.21, s | 17.3 | 1.32, s | 14.8 | 0.82, s |
- —National Natural Science Foundation of China10.13039/501100001809
- —State Administration of Traditional Chinese Medicine of the People's Republic of China10.13039/501100005891
- —Anhui University of Chinese Medicine10.13039/501100009004
- —Key Technologies Research and Development Program of Anhui Province10.13039/100016073
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhytochemical compounds biological activities · Natural product bioactivities and synthesis · Biological Activity of Diterpenoids and Biflavonoids
Introduction
The plant genus Munronia (Meliaceae) comprises approximately 15 species, primarily distributed in China, Sri Lanka, India, Indonesia, and the Philippines. Among them, about 7–8 species native to China and is traditionally utilized in folk medicine, particularly among ethnic minorities.^1^ The whole herb of Munronia is widely employed to treat malaria, rheumatic joint pain, coughs, stomachaches, and has demonstrated notable insecticidal activity.^2^ Although the medicinal value of Munronia plants has been documented in numerous ethnic medical classics, scientific data on their chemical composition and pharmacological effects remain limited. Current studies reveal that Munronia species are rich in limonoids and triterpenoids,^2–5^ which exhibit notable biological activities, including anti-inflammatory,^6^ anti-tumor,^2^ anti-TMV (tobacco mosaic virus),^4^ and anti-proliferative effects.^5^ Our research group previously isolated 74 limonoids, 4 triterpenoids, and 2 pregnanes from Munronia unifoliolata Oliv.^6–8^ Among these, munronoid I demonstrated significant anti-inflammatory activity by markedly suppressing IL-1β release through inhibition of NLRP3 inflammasome initiation and assembly.^6^ To identify additional anti-inflammatory compounds from Munronia species, we performed a systematic investigation of the chemical constituents of Munronia henryi and screened them for anti-inflammatory activity. This study led to the isolation of three pimarane-type diterpenoids from M. henryi.
Pimarane diterpenoids are characterized by a 4, 4, 10, 13-tetramethylperhydrophenanthrene core skeleton and are classified into pimarane, isopimrane, ent-pimarane and ent-isopimrane based on their stereochemical configurations.^9^ These compounds primarily occur in plants from the Lamiaceae, Zingiberaceae, and Cupressaceae families, as well as in fungi and marine organisms. To date, over 360 pimarane-type molecules have been reported, demonstrating notable cytotoxicity, anti-inflammatory, and antimicrobial activities.^10^ However, such diterpenoids are rarely documented in Meliaceae plants, with only minor quantities isolated from Dysoxylum, Guarea, and Chukrasia species.^11^ This study reports, for the first time, the presence of pimarane-type diterpenoids in Munronia plants, including compound 1, which features a unique 6,7-seco pimarane skeleton (Fig. 1). Additionally, their cytotoxic and anti-inflammatory activities are evaluated.
The structures of compounds 1–4.
Results and discussion
Compound 1 was obtained as a colorless square crystal, with a molecular formula of C_20_H_30_O_4_, as established by HR-ESI-MS (m/z 357.2046 [M + Na]^+^, calcd 357.2036), indicative of six degrees of unsaturation. The ^1^H NMR spectrum (Table 1) showed the presence of four olefinic protons (δH 6.33, 1H, s; 5.73, 1H, dd, J = 17.4, 10.6 Hz; 4.96, 1H, d, J = 10.6 Hz; 4.82, 1H, d, J = 17.4 Hz), and four tertiary methyl groups (δH 1.21, 1.16, 1.14 and 1.03, each 3H, s). Combined analysis of the ^13^C NMR and HSQC spectra (Table 1) exhibited 20 carbon signals, including four methyls, six methylenes (one olefinic at δC 112.7), four methines (two olefinic at δC 147.1, 144.7), six quaternary carbons (two carboxyls at δC 177.1, 174.9 and one olefinic at δC 135.3). The above data suggest that compound 1 contains one trisubstituted double bond, one terminal double bond, and four tertiary methyl groups, which are the characteristic fragments of pimarane-type diterpenoid.^12^ However, the six degrees of unsaturation of compound 1 (accounting for two double bonds and two carboxyl groups) imply the presence of only two rings in its structure. Additionally, the two carboxyl groups indicate that the pimarane skeleton of compound 1 has undergone ring-opening.
The 2D NMR spectra revealed key structural features of compound 1 (Fig. 2). In the HMBC spectrum, H_3_-18 (δH 1.03, 3H, s) showed correlations to C-19 (δC 24.5), C-3 (δC 42.0), C-4 (δC 34.5), and C-5 (δC 58.6). These correlations, together with the ^1^H–^1^H COSY cross-peaks between H-1/H-2/H-3, confirmed the intact nature of ring A. Furthermore, HMBC correlations of H_3_-17 (δH 1.14, s) to C-12 (δC 33.8) and C-14 (δC 144.7), along with those of H-14 (δH 6.33, s) to C-9 (δC 41.6) and C-12, combined with the ^1^H–^1^H COSY cross-peaks of H-9/H-11/H-12, established the complete ring C structure. Furthermore, the HMBC correlations between H-14 and δC 174.9, and between H-5 and δC 177.1 suggested that the two carboxyl groups belong to C-7 and C-6, respectively. This assignment was further supported by the molecular formula derived from HR-MS analysis. Consequently, compound 1 was identified as a 6,7-seco pimarane-type diterpenoid, as illustrated in the Fig. 1. Notably, the ^13^C NMR spectrum of 1 exhibits signal broadening, likely attributable to steric hindrance from the carboxyl group restricting free rotation about the C9–C10 single bond.^13^
Key 2D NMR correlations of 1.
The relative configuration of compound 1 was partially established through ROESY correlations (Fig. 2). The cross-peaks between H_3_-18/H-5/H-3α indicated α-orientation for both H_3_-18 and H-5, while the correlation between H_3_-19 and H-3β supported β-orientation for H_3_-19. However, the stereoconfiguration of compound 1 could not be determined by ROESY due to the free rotation about the C9–C10 single bond. The absolute configuration was ultimately determined by X-ray crystallographic analysis of single crystals (Fig. 3) obtained from methanol solution, which established the 5S, 9R, 10R, 13R configuration [Flack parameter = −0.06(13)].
X-ray structure of 1.
Compound 2 was obtained as a white powder. Its molecular formula was established as C_20_H_28_O_3_ by HR-ESI-MS (m/z 315.1960 [M − H]^−^), corresponding to seven degrees of unsaturation. The ^1^H NMR spectrum of 2 (Table 1) displayed signals characteristic of a terminal double bond (δH 5.75, dd, J = 17.6, 10.9 Hz; 5.04, d, J = 10.9 Hz; 4.93, d, J = 17.6 Hz) and four tertiary methyl groups (δH 1.32, 1.16, 0.93 and 0.89, each 3H, s). The ^13^C spectrum of 2, combined with the HSQC spectrum, revealed the presence of seven quaternary carbons (including two keto carbonyls at δC 204.1 and 200.3, and two olefinic carbons at δC 156.0 and 141.4), three methine carbons (one olefinic at δC 143.9, and one oxygenated at δC 69.0), six methylene carbons (one olefinic at δC 113.9) and four methyl carbons. These signals account for four degrees of unsaturation, while the remaining three unsaturations suggest that 2 is a tricyclic diterpenoid. The spin systems identified in the ^1^H–^1^H COSY spectrum (Fig. 4), specifically H-1/H-2/H-3 and H-5/H-6, along with key HMBC correlations (H_3_-18 to C-3/C-4/C-5; H-20 to C-1/C-5/C-9/C-10; and H-6 to C-7/C-8/C-10), were crucial for establishing the structures of rings A and B. Combined with the terminal double bond feature and HMBC correlations from H-17 to C-12, C-14, C-15, compound 2 was identified as a pimarane-type diterpenoid, exhibiting a planar structure similar to known compound 12β-Hydroxy-7,11-dioxopimal-8,15-dien.^14^ The sole structural difference involves the position of hydroxyl group substitution in 2, the hydroxyl group is located at C-14, whereas in 12β-hydroxy-7,11-dioxopimar-8,15-dien, it is positioned at C-12. This distinction was confirmed by the HMBC correlations observed between H-14 and C-7/C-8.
Key 2D NMR correlations of 2.
The ROESY spectrum revealed key spatial relationships: correlations between H_3_-19 (δH 0.93), H-6β (δH 2.44), H_3_-20 (δH 2.69) indicated their cofacial arrangement and β-orientation. Similarly, the H_3_-18 (δH 0.89)/H-6α (δH 2.56) correlation suggested their cofacial α-orientation (Fig. 4). The α-orientation of H-5 was further supported by its small coupling constant (J5,6α = 3.2 Hz). While the ROESY correlation between H_3_-17(δH 1.16) and H-14 (δH 4.55) confirmed their cofacial arrangement, two possible relative configurations (13S**, 14R** and 13R**, 14S**) remained. To resolve this ambiguity, we calculated the ^13^C chemical shifts for both epimers (Fig. 5A). DP4+ analysis of both ^1^H and ^13^C NMR data unequivocally identified (13S**, 14R**)-2 as the correct structure (100% probability, Fig. S3†).^15,16^ The absolute configuration (5S, 10S, 13S, 14R) was ultimately confirmed by excellent agreement between experimental and calculated ECD spectra (Fig. 5B).
Correlations between experimental and calculated 13C NMR chemical shifts of (13S, 14R*)-2 (A) and ECD calculations for (13S, 14R)-2 (B).*
Compound 3 was isolated as a white powder. Its molecular formula (C_20_H_32_O_2_) was established by ^13^C NMR and HR-ESI-MS (observed [M − H]^−^ at m/z 327.2295), implying five degrees of unsaturation. The ^1^H NMR spectrum (Table 1) displayed characteristic methyl group signals (δH 1.08, 0.91, 0.87, 0.82, each, 3H, s) and olefinic proton signals (δH 5.80, dd, J = 17.4, 10.8 Hz; 5.54, d, J = 2.0 Hz; 5.15, d, J = 10.8 Hz; 5.14, d, J = 17.4 Hz), suggesting a pimarane-type diterpenoid skeleton analogous to compound 2. Comparative analysis of 1D and 2D NMR data revealed close structural similarity to (5S, 9R, 10S, 12R, 13R)-12-hydroxyisopimara-8(14),15-dien-7-one,^17^ with only differences of the absence of a carbonyl carbon and the presence of an additional oxygenated carbon. The observed mass difference of 2 suggested reduction of the C-7 carbonyl group to a hydroxyl group in compound 3. This structural modification was confirmed by HMBC correlations from H-7 (δH 3.99) to C-5 (δC 52.0), C-8 (δC 139.4), C-9 (δC 50.3), along with ^1^H–^1^H COSY cross-peaks between H-5/H-6/H-7 (Fig. 6).
Key 2D NMR correlations of 3.
ROESY correlations observed among H_3_-18/H-5/H-9/H-12, H-5/H-7, and H_3_-18/H-6α (δH 2.00) established their cofacial arrangement and α-orientation. Complementary ROESY cross-peaks between H_3_-17/H-11β (δH 1.53)/H_3_-20/H-6β (δH 1.29) confirmed their β-orientation. The absolute configuration of 3 was determined to be 5S, 7S, 9R, 10S, 12R, 13R through comparison of experimental and calculated ECD spectra (Fig. S2†).
In addition to the aforementioned compounds, a known pimarane-type diterpenoid was isolated from M. henryi. Based on comparison of its spectroscopic data with literature values,^17^ this compound was identified as (5S, 9R, 10S, 12R, 13R)-12-hydroxyisopimara-8(14),15-dien-7-one (4).
According to previous literature, pimarane-type diterpenoids have demonstrated significant cytotoxic and anti-inflammatory potential.^18–20^ Initially, all compounds were tested for anti-inflammatory activity using L-NMMA (50 μM) as a positive control (52.75 ± 1.28% inhibition). At 50 μM concentration, compounds 1–4 exhibited no significant inhibitory effects on NO production in LPS-induced RAW 264.7 macrophages, showing inhibition rates ranging from 13.73% to 32.35%.
Subsequently, the cytotoxicity of these compounds was evaluated against human colon cancer cells (HCT-166) and human liver cancer cells (Hep3B). None of the compounds exhibited significant cytotoxicity against either cancer cell line.
Conclusions
In summary, four pimarane-type diterpenoids were isolated from the petroleum ether extract of Munronia henryi, including three previously undescribed compounds (1–3) and one known analogue. Muhenrin A (1) features a novel 6,7-seco pimarane skeleton, likely biosynthesized from 7α-hydroxyisopimara-8(14),15-diene.^21^ Two possible pathways are proposed for its formation (Scheme S1†). Path a: the 7-hydroxyl group of 7α-hydroxyisopimara-8(14),15-diene is oxidized to a ketone carbonyl (i), followed by Baeyer–Villiger oxidation to yield the lactone intermediate (ii).^22^ Subsequent hydrolysis and continuous oxidation produce 1. Path b: 7α-hydroxyisopimara-8(14),15-diene undergoes dehydration to form the alkene intermediate (iv), which is then directly oxidized to 1via dioxygenase activity.^23^ To our knowledge, this represents the first report of pimarane-type diterpenoids from Munronia species.
The initial biological activity assays did not yield significant results. Future studies should consider alternative cell models or explore additional biological activities, such as antibacterial or antiviral effects.^24,25^
Experimental
General experimental procedures
Optical rotations were measured with a P-2000 digital polarimeter (solvent: MeOH, JASCO, Japan). UV spectra were obtained on a UV-2401A spectrophotometer (solvent: MeOH, Shimadzu, Japan). Circular dichroism (CD) spectra were obtained on a JASCO(J-1500) CD spectrometer in MeOH (Applied Photophysics, Japan). HRMS-ESI was performed on an Thermo Orbitrap Exploris 120 mass spectrometer (Thermo Fisher Scientific, USA, Xevo G2 - XSQT of mass spectrometer from Waters, USA). In addition, 1D and 2D NMR spectra were recorded on Bruker DRX-500 and 600 spectrometers (Bruker, Germany) at 298 K (internal standard: TMS, solvent: CD_3_OD and CDCl_3_). Silica gel (200–300 mesh, Qingdao Haiyang, China), Sephadex LH-20 (GE, USA), and RP-18 (5 μm, Fuji Silysia Chemical, Japan) were used for column chromatography. MPLC was performed on a Büchi Sepacore System equipped with a pump manager (Büchi, Switzerland) and prep-HPLC was performed on a 1260 instrument (Agilent, USA).
Plant material
The whole plant of Munronia henryi was collected in November 2022 from Wenshan Zhuang and Miao Autonomous Prefecture, Yunnan province, P. R. China, and identified by Professor Qing-Shan Yang of Anhui University of Chinese Medicine. A voucher specimen (no. 20221101) was deposited in the Department of Traditional Chinese Medicine and Natural Medicinal Chemistry at Anhui University of Chinese Medicine.
Extraction and isolation
The dried and pulverized aerial parts of Munronia henryi (20 kg) were exhaustively extracted with 95% ethanol (4 × 3 h, reflux). The combined ethanolic extracts were concentrated under reduced pressure to yield a crude extract, which was subsequently suspended in H_2_O and partitioned with dichloromethane (DCM). The DCM portion was suspended in water and then extracted with petroleum ether (PE).
The PE fraction (363 g) was fractionated by silica gel column chromatography using a stepwise gradient of PE-EtOAc (100 : 0 → 0 : 100, v/v) to yield eight fractions (A–H). Fraction F (27.1 g) was further purified by ODS column chromatography with a MeOH–H_2_O gradient (40 : 60 → 100 : 0, v/v), yielding six subfractions (Fa–Ff). Subfraction Fc was chromatographed on ODS with isocratic elution (MeCN–H_2_O, 45 : 55, v/v) to afford nine fractions (Fca–Fci). Final purification of Fcc by semi-preparative HPLC (MeCN–H_2_O, 65 : 35, 8.0 mL min^−1^) yielded compound 4 (4.81 mg, tR = 36 min).
Similarly, subfraction Fb was separated by ODS chromatography (isocratic MeCN–H_2_O, 45 : 55) into thirteen fractions (Fba–Fbn). Fraction Fbk was purified by semi-preparative HPLC (MeCN– H_2_O 57 : 43, 8.0 mL min^−1^) to give Fbka (4.71 mg, tR = 43 min), which was further purified (MeOH–H_2_O, 75 : 25, 8.0 mL min^−1^) to afford compound 3 (2.58 mg, tR = 28 min). Fraction Fbn was processed similarly (MeCN–H_2_O, 67 : 33 → MeOH–H_2_O, 55 : 45) to yield compound 2 (2.55 mg, tR = 32 min) via intermediate fraction Fbna (5.31 mg, tR = 45 min).
Fraction H (24.2 g) was fractionated by MCI gel column chromatography using a MeOH–H_2_O gradient (20 : 80 → 100 : 00, v/v), yielding six subfractions (Ha–Hf). Subfraction Hf was further purified by Sephadex LH-20 column chromatography (MeOH) to afford four fractions (Hfa–Hfd). Final purification of Hfb by semi-preparative HPLC (MeCN–H_2_O gradient: 0–30 min, 55 : 45; 30–35 min, 55 : 45 → 85 : 15; 35–50 min, 85 : 15; flow rate 8.0 mL min^−1^) yielded compound 1 (6.75 mg, tR = 43 min).
Muhenrin A (1)
Colorless square crystal; [α]25 D − 53.0 (c = 0.2, MeOH); UV λmax (MeOH) nm (log ε): 210 (4.62); ECD (MeOH) λmax (Δε) 237.5(+16.72) nm; IR (KBr) cm^−1^ 3457, 2951, 2864, 2831, 1701, 1604, 1448, 1365, 1227, 1205, 1152; ^1^H and ^13^C NMR data see Table 1; HRESI-MS m/z 357.2046[M + Na]^+^ (calcd for 357.2036, C_20_H_30_O_4_Na).
Muhenrin B (2)
White powder; [α]25 D − 2.9 (c 0.31, MeOH); UV (MeOH) λmax (log ε) 239 (2.24) nm; ECD (MeOH) λmax (Δε) 205(−8.87) nm, 217 (−2.92) nm, 242 (−7.5) nm, 279 (+8.9) nm; IR (KBr) cm^−1^ 3394, 2922, 2831, 2716, 1600,1364, 1154, 1068, 971,775; ^1^H and ^13^C NMR data see Table 1; HR-ESI-MS m/z 315.1960 [M − H]^−^ (calcd for 315.1955, C_20_H_27_O_3_).
Muhenrin C (3)
White power; [α]25 D + 10.3 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 210 (3.12) nm; ECD (MeOH) λmax (Δε) 299 (+0.48) nm; IR (KBr) cm^−1^ 3394, 2922, 2831, 2716, 1600,1364, 1154, 1068, 971,775; ^1^H and ^13^C NMR data see Table 1; HR-ESI-MS m/z 327.2295[M + Na]^+^(calcd for 327.2294, C_20_H_32_O_2_Na).
Crystal data for muhenrin A (1)
C_20_H_30_O_4_, M = 334.44, a = 12.0825(8) Å, b = 12.7004(8) Å, c = 12.9052(8) Å, α = 90°, β = 90°, γ = 90°, V = 1980.3(2) Å^3^, T = 170.0 K, space group P2_1_, Z = 4, μ(Cu Kα) = 0.613 mm^−1^, 17 312 reflections measured, 3988 independent reflections (Rint = 0.0736). The final R1 values were 0.0582 (I > 2σ(I)). The final wR(F^2^) values were 0.1559 (I > 2σ(I)). The final R1 values were 0.0621 (all data). The final wR(F^2^) values were 0.1622 (all data). The goodness of fit on F^2^ was 1.019. Flack parameter = −0.06(13). A suitable crystal was selected and recorded with a diffractometer using Cu Kα radiation. The structure was solved with the ShelXT structure solution program using direct methods and refined with the ShelXL refinement package using least squares minimisation based on Olex2 software. The crystallographic data of compounds have been deposited at the Cambridge Crystallographic Data Center with the deposition number CCDC 2452064 (1).
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
RA-015-D5RA04525H-s001
RA-015-D5RA04525H-s002
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Flora Reipublicae Popularis Sinicae, ed. Editorial Committee of FRPS, Science Press, Beijing, 1997, vol. 43(3), p. 57
- 2Yan Y. Zhang J.-X. Huang T. Mao X.-Y. Gu W. He H.-P. Di Y.-T. Li S.-L. Chen D.-Z. Zhang Y. Hao X.-J. J. Nat. Prod.20157848118212579852810.1021/np 501057 f · doi ↗ · pubmed ↗
- 3Yang X.-R. Tanaka N. Tsuji D. Lu F.-L. Yan X.-J. Itoh K. Li D.-P. Kashiwada Y. Tetrahedron 201975130779
- 4Yan Y. Wang D. Zhang X. Peng M.-Y. Yan X.-Y. Guo Y.-S. Jia M.-G. Zhou J. Tang L. Hao X.-J. Pestic. Biochem. Physiol.20221841051083571504710.1016/j.pestbp.2022.105108 · doi ↗ · pubmed ↗
- 5Yang X.-R. Liu P.-y. Wei Y.-L Song J.-R. Yan X.-J. Jiang X.-H. Li J.-X. Li X.-Q. Li D. P. Lu F.-L. Molecules 20232868393783668110.3390/molecules 28196839 PMC 10574247 · doi ↗ · pubmed ↗
- 6Sun Y.-P. Li Q.-R. Cui L.-T. Tang P.-F. Li Y.-Y. Kong L.-Y. Luo J. Chin. J. Chem.202240123136
- 7Sun Y.-P Cui L.-T. Li Q.-R. Tang P.-F Li Y.-Y. Xu W.-J. Luo J. Kong L. Y. Chin. Chem. Lett.202233516518
- 8Sun Y.-P. Li Q. R. Sun Y. J. Cui L. T. Wang Y.-Y. Li Y.-Y. Luo J. Kong L. Y. J. Nat. Prod.202184235223653435775510.1021/acs.jnatprod.1c 00519 · doi ↗ · pubmed ↗