Complete genome sequence data of Xylella fastidiosa subspecies multiplex ST88 and ST89 indicate distinct introductions in France
Amandine Cunty, Jessica Dittmer, Déborah Merda, Bruno Legendre, Benoit Remenant, Yannick Blanchard, Sophie Cesbron, Marie-Agnès Jacques, Pascal Gentit, Anne-Laure Boutigny

TL;DR
This paper reports the complete genome sequences of two new Xylella fastidiosa strains in France, helping to understand their origin and diversity.
Contribution
The study provides complete genome sequences and phylogenetic analysis of two new Xylella fastidiosa subspecies multiplex strains (ST88 and ST89) in France.
Findings
ST88 and ST89 strains were isolated from infected plants in Provence-Alpes-Côte d’Azur.
Genome assemblies were created using PacBio and Illumina sequencing data.
Phylogenomic analysis revealed distinct introductions of these strains in France.
Abstract
Xylella fastidiosa is a Gram-negative bacterium native to the Americas and classified as a priority pest under EU regulations. This xylem-limited plant pathogenic bacterium has a wide host range and is transmitted by insect vectors. Since 2013, X. fastidiosa has been identified in several European countries including Italy, France, Spain and Portugal, with different subspecies and sequence types (ST) detected. Since 2015, most strains identified in France are of the subspecies multiplex, specifically ST6 and ST7. Two new STs of X. fastidiosa subsp. multiplex, ST88 and ST89, were recently detected in the region Provence-Alpes-Côte d’Azur (PACA), and one strain of each ST has been isolated from infected plants. To investigate the phylogenetic relationships between the four STs present in France, a complete circular genome and a single-contig genome were assembled for the ST89 and ST88…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
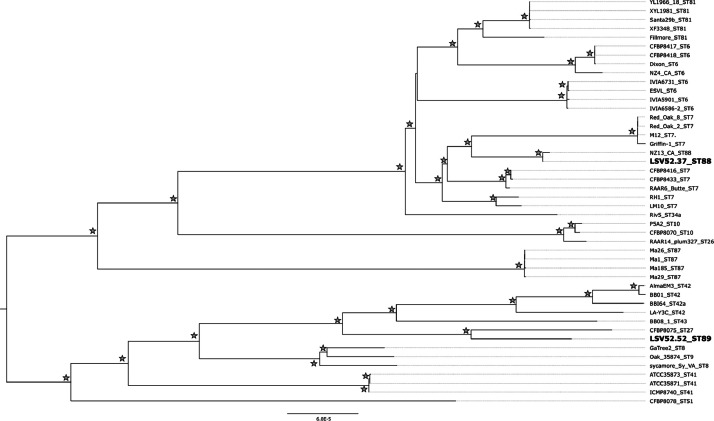 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhytoplasmas and Hemiptera pathogens · Mycorrhizal Fungi and Plant Interactions · Cocoa and Sweet Potato Agronomy
Specifications TableSubjectMicrobiologySpecific subject areaBacteriology, Fastidious bacteria, GenomicsType of dataTable, FigureGenomic data, Raw, Analyzed, Filtered, ProcessedData collectionGenomic DNA extraction: Wizard Genomic DNA Purification Kit (Promega) and QuickPick^TM^ SML Plant DNA kit (Bio-Nobile)Genome sequencing platforms: Illumina MiSeq and PacBioBioinformatic tools: Canu v2.1.1, Flye v2.8.1, Circlator v1.5.6, Polca from the MaSuRCa toolkit v4.0.9, CheckM v1.1.6, Bakta v1.5.1Data source locationPlant Health Laboratory of ANSES, Angers, FranceIRHS, Angers, FranceData accessibilityRepository name: NCBI (www.ncbi.nlm.nih.gov)Data identification number: Bioprojects PRJNA1234384 and PRJNA1234386Direct URL to data:LSV 52.37:https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1234384https://www.ncbi.nlm.nih.gov/biosample/SAMN47296926https://www.ncbi.nlm.nih.gov/sra/SRX27952896https://www.ncbi.nlm.nih.gov/sra/SRX27952897https://www.ncbi.nlm.nih.gov/sra/SRX27952898LSV 52.52:https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1234386https://www.ncbi.nlm.nih.gov/biosample/SAMN47297024https://www.ncbi.nlm.nih.gov/sra/SRX27953175https://www.ncbi.nlm.nih.gov/sra/SRX27953176https://www.ncbi.nlm.nih.gov/sra/SRX27953177Instructions for accessing these data: The complete genome sequences of Xylella fastidiosa subsp. multiplex strains LSV52.37 (ST88) and LSV52.52 (ST89) are available in the National Centre for Biotechnology Information (NCBI) database (Bioproject PRJNA1234384, Biosample SAMN47296926 for LSV52.37 and Bioproject PRJNA1234386, Biosample SAMN47297024 for LSV52.52). The raw data are available under the following accession numbers: SRR32648475 to SRR32648477 for LSV52.37 and SRR32648754 to SRR32648756 for LSV52.52.Related research articleA. Cunty, B. Legendre, P. de Jerphanion, C. Dousset, A. Forveille, S. Paillard, V. Olivier, Update of the Xylella fastidiosa outbreak in France: two new variants detected and a new region affected, Eur. J. Plant Pathol. 163 (2022) 505–510. 10.1007/s10658-022-02492-z.
Value of the Data
1
- •Data report on the high-quality genome sequences of two strains of X. fastidiosa subsp. multiplex ST88 and ST89 isolated in France.
- •The data extend genomic data available for European strains of X. fastidiosa subsp. multiplex and are valuable for comparative genomics.
- •The data provide a better overview of the diversity and origin of X. fastidiosa subsp. multiplex in France and Europe.
- •The data support the hypothesis of two distinct introductions from the USA to France for both ST88 and ST89, rather than both ST88 and ST89 having evolved from strains already present in France. These results are in accordance with the MLST analysis previously performed on these two strains [1].
Background
2
Xylella fastidiosa is a Gram-negative, xylem-limited phytopathogenic bacterium with a broad host range, which causes significant epidemics and economic losses in the Americas and Europe [2]. Despite being native to the Americas [3], since 2013 X. fastidiosa has been identified in various plant species in European countries with a Mediterranean climate, such as Italy, France, Spain and Portugal, with different subspecies and sequence types (ST) detected [2,4]. These detections are the result of multiple independent introductions likely originating from the USA [4,5]. In France, the subspecies multiplex was first detected in 2015 in Corsica and in the region Provence-Alpes-Côte d’Azur (PACA) (ST6 and ST7) and in 2020 in the Occitanie region (ST6) [1,6]. Also in 2020, two new STs of X. fastidiosa subsp. multiplex were identified in the PACA region at two different locations and in different host plants: (i) ST88 was detected on Polygala myrtifolia, Hebe sp., Osteospermum ecklonis, Lavandula x intermedia, Coronilla glauca and Euryops chrysanthemoides and (ii) ST89 was detected on Myoporum sp. and Viburnum tinus [1]. Here, we report the high-quality genome sequence of an ST88 strain and the complete circular genome sequence of an ST89 strain by combining PacBio and Illumina sequencing data. In addition, we performed a phylogenomic analysis of the subspecies multiplex to investigate the phylogenetic position and potential origin of these new strains.
Data Description
3
Long- and short-reads were obtained via the PacBio HiFi and Illumina platforms, respectively. The final assemblies resulted in a single non-circularized contig for the ST88 strain LSV52.37 and a complete circular genome for the ST89 strain LSV52.52 (Table 1). No plasmids were detected in either genome. The genome of the ST88 strain was longer than the genome of the ST89 strain (2.69 Mbp and 2.52 Mbp, respectively) but both genomes are in the typical size range for X. fastidiosa genomes. The number of pseudogenes was very low (25 and 32, respectively) and CheckM confirmed a high genome completeness and absence of contamination (Table 1). The genome assemblies and the raw sequencing data were deposited in the NCBI databases under BioProject numbers PRJNA1234384 for X. fastidiosa subsp. multiplex ST88 (strain LSV52.37) and PRJNA1234386 for X. fastidiosa subsp. multiplex ST89 (strain LSV 52.52). A phylogenomic analysis based on SNPs was performed using the two genomes obtained in this study and 44 genome sequences representing the genetic diversity of the subspecies multiplex available in GenBank as of 12/12/2024 (Table 2, Fig. 1).Table 1. Genome assembly and annotation metrics of the two Xylella fastidiosa subsp. multiplex ST88 and ST89 strains.Table 1. MetricsLSV52.37 (ST88)LSV52.52 (ST89)Nb of reads (PacBio/Illumina)56,996 / 3,967,32869,832 / 3,895,010Genome size (bp)2,698,4302,524,893Nb of contigs11GC (%)51.7751.81Coverage (long reads)171243Coverage (short reads)282256CDS26792409Pseudogenes2532rRNA66tRNA5554tmRNA11ncRNA96Regulatory ncRNA22CheckM Completeness (%)99.2899.64CheckM Contamination (%)0.000.00BioProjectPRJNA1234384PRJNA1234386Table 2Genome sequences of X. fastidiosa subsp. multiplex strains available in GenBank used for the phylogenomic analysis.Table 2. Strain nameSTHost plantLocationAccessionCFBP84186Spartium junceumFrance: CorsicaGCF_042244175.1CFBP84176Spartium junceumFrance: CorsicaGCF_042244685.1Dixon6Prunus dulcisUSA: CaliforniaGCF_029626005.1ESVL6Prunus dulcisSpain: AlicanteGCF_004023385.1IVIA59016Prunus dulcisSpain: AlicanteGCF_004023395.2IVIA6586-26Helichrysum italicumSpain: AlicanteGCF_009669335.1IVIA67316Helichrysum italicumSpain: AlicanteGCF_009669375.1NZ4_CA6Brachyglottis compactaUSA: CaliforniaGCF_043355825.1CFBP84167Polygala myrtifoliaFrance: CorsicaGCF_001971475.1CFBP84337Cistus monspeliensisFrance: CorsicaGCF_028752655.1Griffin-17Quercus rubraUSA: GeorgiaGCA_000466025.1LM107Olea europaeaUSA: CaliforniaGCF_012974145.1M127Prunus dulcisUSA: CaliforniaGCF_000019325.1RAAR6 Butte7Prunus dulcisUSA: CaliforniaGCF_009695485.1Red Oak 27Quercus rubraUSA: GeorgiaGCF_015475935.1Red Oak 87Quercus rubraUSA: GeorgiaGCF_021459885.1RH17Olea europaeaUSA: CaliforniaGCF_012974125.1sycamore Sy-VA8Platanus occidentalisUSA: VirginiaGCF_000732705.1GaTree28Carya illinoinensisUSA: GeorgiaGCA_042862445.1Oak 358749Quercus sp.USA: WashingtonGCF_021459905.1CFBP807010Prunus sp.USA: GeorgiaGCF_042243345.1P5A210Prunus persicaUSA: AlabamaGCA_022548865.1RAAR14 plum32726Prunus domesticaBrazil: Rio Grande do SulGCF_009695495.1CFBP807527Prunus sp.USA: CaliforniaGCF_042242545.1Riv534Prunus cerasiferaUSA: CaliforniaGCF_015475955.1CFBP8173 (ATCC 35871)41Prunus salicinaUSA: GeorgiaGCF_042241145.1ICMP874041Platanus occidentalisUSA: WashingtonGCF_028735895.1CFBP8068 (ATCC 35873)41Ulmus sp.USA: WashingtonGCF_042241895.1AlmaEm342Vaccinium sp.USA: GeorgiaGCF_018069645.1BB0142Vaccinium corymbosumUSA: GeorgiaGCF_001886315.1LA-Y3C42Vaccinium virgatumUSA: LouisianaGCF_021459845.1BBI6442Vaccinium sp.USA: GeorgiaGCA_006369955.1BB08-143Vaccinium ``Windsor''USA: FloridaGCF_018069665.1CFBP807851Vinca sp.USA: FloridaGCF_004016365.1Fillmore81Olea europaeaUSA: CaliforniaGCF_012974105.1XF334881Prunus dulcisSpain: MajorcaGCF_042239545.1XYL1966/1881Olea europaeaSpain: MinorcaGCF_042238405.1XYL198181Ficus caricaSpain: MajorcaGCF_009669455.1Santa29b81Santolina chamaecyparissusSpain: MinorcaGCF_042240315.1Ma187Rhamnus alaternusItaly: TuscanyGCF_018449155.1Ma2687Spartium junceumItaly: TuscanyGCF_018449175.1Ma2987Prunus dulcisItaly: TuscanyGCF_018449135.1Ma18587Polygala myrtifoliaItaly: TuscanyGCF_018449105.1NZ13_CA88Pomaderris prunifoliaUSA: CaliforniaGCF_043195255.1Fig. 1Phylogenomic tree showing the relationships of X. fastidiosa subsp. multiplex strains using maximum likelihood analysis. The tree was visualized using FigTree. Strains sequenced in this study are in bold. Bootstraps > 80% are represented by grey stars.Fig 1
This analysis revealed that the ST88 strain (LSV52.37) isolated from Polygala myrtifolia in France clustered with another ST88 strain (NZ13_CA) recently isolated from Pomaderris prunifolia in California (USA) [7]. The two ST88 strains formed a highly-supported clade most closely related to ST7 strains from Red Oak and almond isolated in the USA. In contrast, the ST89 strain (LSV52.52) was closely-related to American ST27 infecting almond and plum.
Experimental Design, Materials and Methods
4
Bacterial Culture
4.1
Xylella fastidiosa subsp. multiplex strain LSV52.37 (ST88) was isolated in 2020 from Polygala myrtifolia in Saint-Raphaël (PACA region) and LSV52.52 (ST89) was isolated in 2021 from Myoporum sp. in Villeneuve-Loubet (PACA region) on modified PWG (Periwinkle Wilt-Gelrite) medium [2]. They are stored at -80°C in the laboratory collection of the ANSES Plant Health Laboratory.
DNA Extraction and Library Preparation
4.2
For PacBio long-read sequencing, bacteria were grown on solid PWG medium at 28°C for seven days. The cultures were replated twice to obtain sufficient bacterial cells. Bacterial cells from these plate cultures were suspended in 10 mL of sterile water with DO_600_ 0.3-0.6. Genomic DNA was extracted from these bacterial suspensions using the Wizard Genomic DNA Purification Kit (Promega, Madison, USA), following the protocol for Gram-positive bacteria with some modifications to improve cell lysis and DNA purity: the initial bacterial cell pellet was washed once in 2 mL of sterile water before being resuspended in 480 µL EDTA (50 mM, pH=8). Lysozyme (120 µL, 10 mg/mL) was then added, followed by incubation at 37°C for 60 min. 35 µL of Pronase (5 mg/mL) was added and the samples incubated overnight at 37°C. Next, 150 µL of 10% SDS (w/v) was added and the samples were incubated at 37°C for 45 min. Nuclei Lysis Solution (800 µL) was then added and the samples were incubated at 80°C for 5 min. Following RNase A treatment at 37°C for 60 min, 270 µL of Protein Lysis Solution was added and incubated on ice for 20 min. After centrifugation at 16,000 g for 6 min, DNA was precipitated in 1 volume of isopropanol, the DNA pellet was washed once with 70% ethanol (v/v) and resuspended in DNA Rehydration Solution. DNA quality was verified using a Nanodrop One (Thermo Fisher Scientific, Waltham, USA) and DNA concentration was quantified using the Qubit dsDNA Broad Range assay kit (Invitrogen, Waltham, USA). Library preparation and sequencing on the PacBio HiFi platform were performed by the Gentyane sequencing facility at Clermont-Ferrand, France. For Illumina sequencing, bacteria were grown on solid modified PWG medium at 28°C for seven days. The cultures were replated twice to obtain sufficient bacterial cells. Bacterial cells from these plate cultures were suspended in 1 mL of sterile water at a concentration of 10^9^ CFU/mL. Genomic DNA was extracted from these bacterial suspensions using the QuickPick^TM^ SML Plant DNA kit (Bio-Nobile, Pargas, Finland). Illumina sequencing libraries were generated as previously described [8].
Sequencing Analysis
4.3
Long-read assemblies were performed using both Canu v2.1.1 [9] and Flye v2.8.1 [10]. For the strain LSV52.37 (ST88), the most contiguous assembly was obtained using Canu with the parameters genomeSize=2.7m, minReadLength=2000, minOverlapLength=1000, producing two contigs. Using the Illumina reads, these two contigs were scaffolded into a single contig with Redundans v0.14 [11] with the parameters –noreduction –limit 1. Circularization was attempted using Circlator v1.5.6 [12] but was not successful. The genome of the strain LSV52.52 (ST89) was directly assembled into a single circular contig using Flye with the parameters –pacbio-hifi, -g 2.7m and –min-overlap 1000. The genome was rotated to start with the gene dnaA using Circlator’s fixstart function. Both assemblies were polished with the Illumina reads using Polca from the MaSuRCa toolkit v4.0.9 [13], but no errors were found, confirming the high quality of the PacBio HiFi assemblies. Genome completeness and contamination was verified using CheckM v1.1.6 [14] and both genomes were annotated using Bakta v1.5.1 [15] (Table 1). Coverage was estimated using Mosdepth [16] as implemented on Galaxy (usegalaxy.org). Default parameters were used for all bioinformatics tools unless stated otherwise.
Phylogenomic Analysis
4.4
Multiple genome alignment of all assemblies (Table 2) was performed using Parsnp v1.5.6 [17]. An in-house python script (supplementary data) was used to concatenate blocks in the xmfa file to obtain a fasta file. Recombination tracts were removed from the alignment using ClonalFrameML v1.12 [18]. The maximum likelihood phylogenetic tree was obtained using IQ-TREE v2.0.3 [19]. The best substitution model was inferred directly by JModelTest [20] implemented in IQ-TREE, and the values of branch support were obtained by bootstrap method using 1000 replicates. The final phylogenetic tree was visualized using FigTree v1.4.4 (https://github.com/rambaut/figtree).
Limitations
None.
Ethics Statement
The authors have read and follow the ethical requirements for publication in Data in Brief. The current work does not involve human subjects, animal experiments, or any data collected from social media platforms
CRediT authorship contribution statement
Amandine Cunty: Conceptualization, Methodology, Writing – original draft. Jessica Dittmer: Methodology, Writing – original draft. Déborah Merda: Formal analysis, Writing – review & editing. Bruno Legendre: Writing – review & editing. Benoit Remenant: Writing – review & editing. Yannick Blanchard: Writing – review & editing. Sophie Cesbron: Methodology, Writing – review & editing. Marie-Agnès Jacques: Funding acquisition, Writing – review & editing. Pascal Gentit: Funding acquisition, Writing – review & editing. Anne-Laure Boutigny: Conceptualization, Methodology, Writing – review & editing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cunty A.Legendre B.De Jerphanion P.Dousset C.Forveille A.Paillard S.Olivier V.Update of the Xylella fastidiosa outbreak in France: two new variants detected and a new region affected Eur. J. Plant Pathol.163202250551010.1007/s 10658-022-02492-z · doi ↗
- 2EPPO, PM 7/24 (5) Xylella fastidiosa EPPO Bull.53202320527610.1111/epp.12923 · doi ↗
- 3Vanhove M.Retchless A.C.Sicard A.Rieux A.Coletta-Filho H.D.De La Fuente L.Stenger D.C.Almeida R.P.P.Genomic diversity and recombination among Xylella fastidiosa subspecies Appl. Environ. Microbiol.85201910.1128/AEM.02972-18e 02972-18PMC 658116431028021 · doi ↗ · pubmed ↗
- 4Landa B.B.Castillo A.I.Giampetruzzi A.Kahn A.Román-Écija M.Velasco-Amo M.P.Navas-Cortés J.A.Marco-Noales E.BarbéS.Moralejo E.Coletta-Filho H.D.Saldarelli P.Saponari M.Almeida R.P.P.in Ea Europe associated with multiple intercontinental introductions Appl. Environ. Microbiol.86202010.1128/AEM.01521-19e 01521-19PMC 697464531704683 · doi ↗ · pubmed ↗
- 5Dupas E.Durand K.Rieux A.Briand M.Pruvost O.Cunty A.DenancéN.Donnadieu C.Legendre B.Lopez-Roques C.Cesbron S.RavignéV.Jacques M.A.Suspicions of two bridgehead invasions of Xylella fastidiosa subsp. multiplex in France Commun. Biol.27202310310.1038/s 42003-023-04499-6PMC 988346636707697 · doi ↗ · pubmed ↗
- 6DenancéN.Legendre B.Briand M.Olivier V.de Boisseson C.Poliakoff F.Jacques M.A.Several subspecies and sequence types are associated with the emergence of Xylella fastidiosa in natural settings in France Plant Pathol.6620171054106410.1111/ppa.12695 · doi ↗
- 7Visnovsky S.B.Kahn A.K.Nieto-Jacobo F.Panda P.Thompson S.Teulon D.A.J.Bojanini Molina I.Virginia Marroni M.Groenteman R.Rigano L.A.Taylor R.K.Forbes H.Almeida R.P.P.Multiple genotypes of a quarantine plant pathogen detected in New Zealand indigenous plants located in a botanical garden overseas Plant Pathol.74202440341210.1111/ppa.14026 · doi ↗
- 8Boutigny A.L.Remenant B.Legendre B.Beven V.Rolland M.Blanchard Y.Cunty A.Direct Xylella fastidiosa whole genome sequencing from various plant species using targeted enrichment J. Microbiol. Methods 208202310671910.1016/j.mimet.2023.10671937028518 · doi ↗ · pubmed ↗